Background: Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) is a rare but life-threatening inherited arrhythmic disorder, typically triggered by exercise or emotional stress, and a leading cause of sudden cardiac death in the young. CPVT is commonly caused by mutations in calcium-handling components of sarcoplasmic reticulum (SR), which promote diastolic calcium leakage and increase arrhythmogenic risk under adrenergic stress. Current therapies such as β-blockers and, in more severe cases, cardiac sympathetic denervation, remain insufficient in a significant subset of patients – suggesting a more complex pathophysiological mechanism. Although previous studies have characterised CPVT phenotypes in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), the contribution of diseased sympathetic neurones (SNs) to the CPVT phenotype remains poorly understood.
Method: hiPSCs derived from both a CPVT patient and a healthy control were differentiated into cardiomyocytes (hiPSC-CMs) and sympathetic neurones (hiPSC-SNs). Electrophysiological activity was assessed using multi-electrode array (MEA) recordings. Calcium transients were measured using Fura-2AM indicator. Real time cAMP dynamics were monitored using a fluorescence resonance energy transfer (FRET) based biosensor (Epac-SH187). All hiPSC lines used in this study were ethically approved and obtained from established sources with appropriate donor consent.
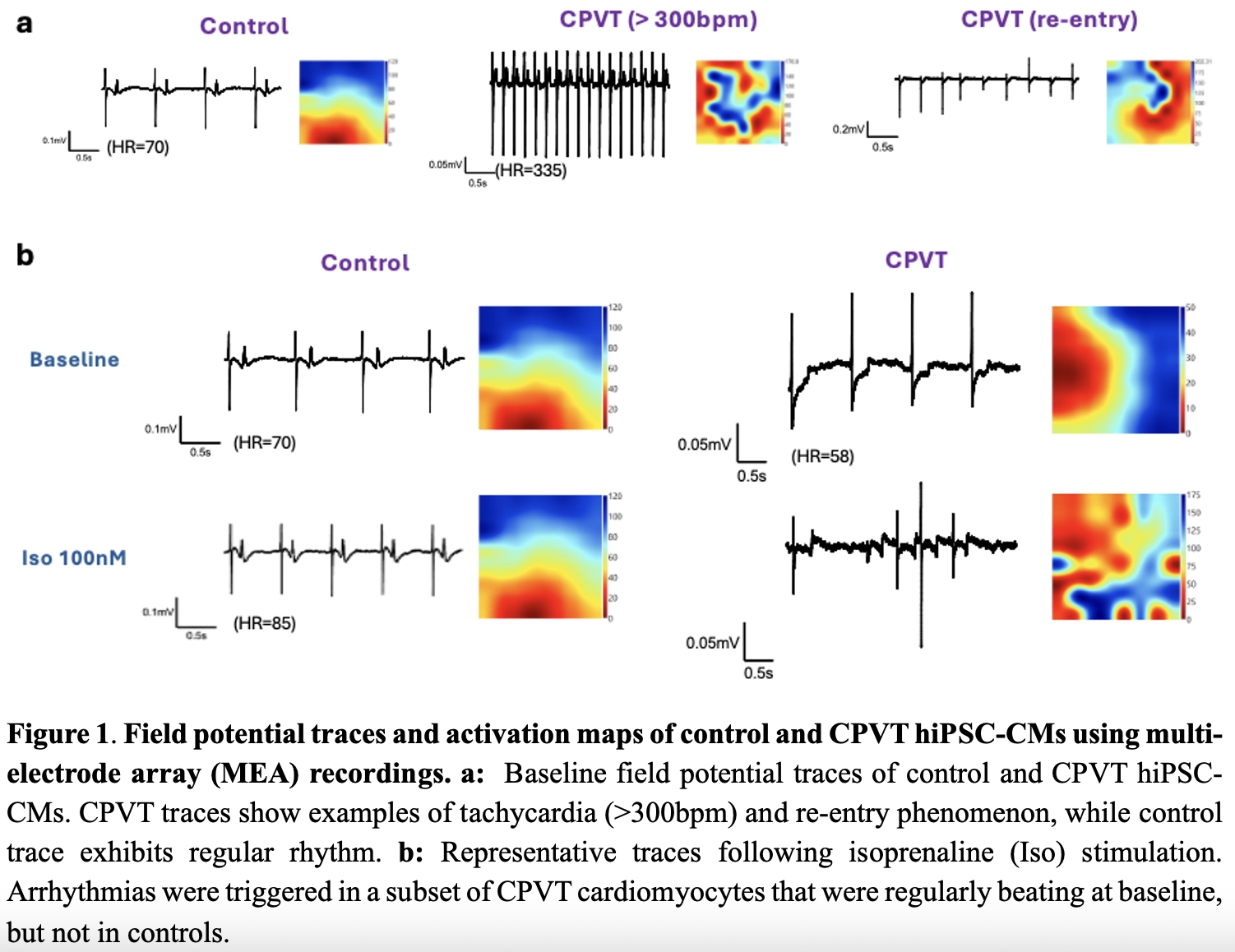
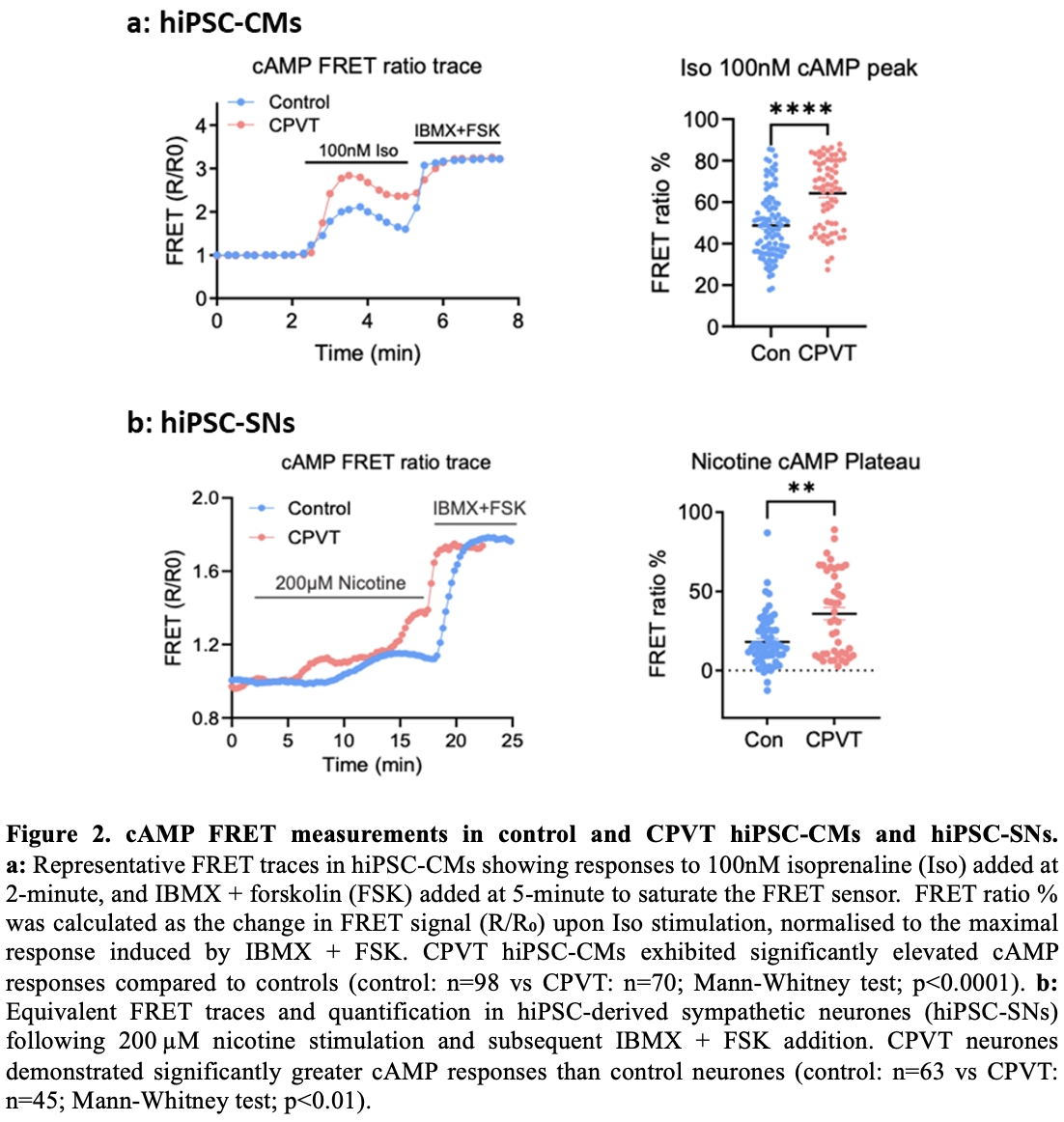
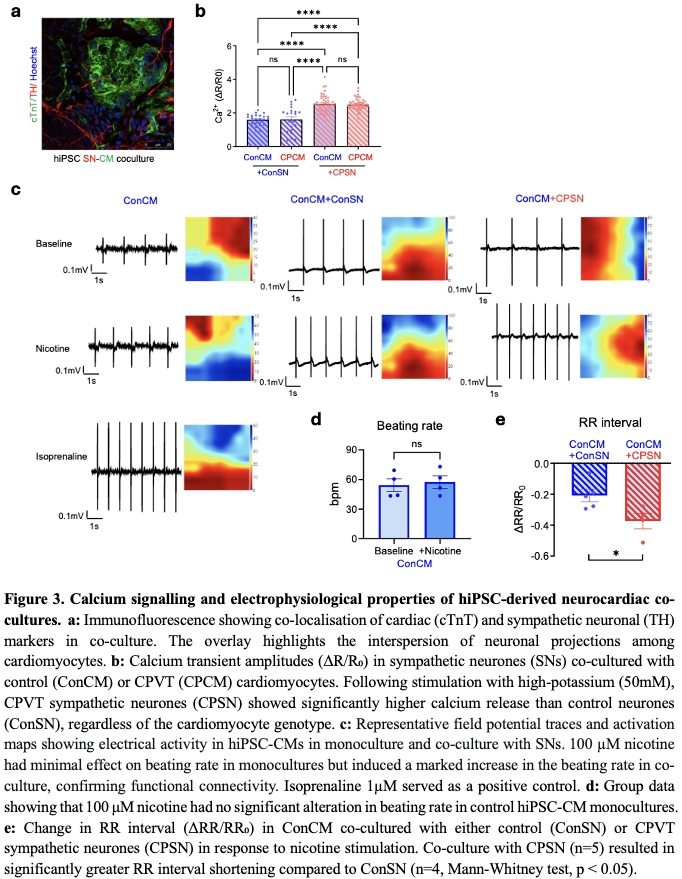
Results: To characterise features of baseline cardiac action potential, MEA measurements were conducted. At baseline, 47% (8 out of 17) of CPVT hiPSC-CMs exhibited spontaneous arrhythmic activity, including tachycardia and re-entry phenomena, compared to none in controls (n=8). Among regularly beating cells, CPVT hiPSC-CMs showed significantly shorter RR intervals (control: 0.5638±0.049s, n=8; vs CPVT: 0.4412±0.025s, n=8; unpaired t-test; p<0.05) and field potential durations (control: 0.3406±0.041s, n=8; vs CPVT: 0.1844±0.015s, n=8; unpaired t-test; p<0.01). Application of isoprenaline (β-adrenergic stimulation) shortened RR interval in both groups. Arrhythmias were triggered in some CPVT cardiomyocytes (regular at baseline), but not in controls. Calcium imaging revealed greater isoprenaline-induced calcium transients in CPVT hiPSC-CM (Control: 0.05±0.01, n=11; vs CPVT: 0.99±0.13, n=14; unpaired t-test, p<0.05). Spontaneous calcium discharges were detected in 77.4% (24 out of 31) of CPVT compared to 10% (1 out of 10) of controls. FRET analysis showed significantly elevated cAMP responses in CPVT hiPSC-CM following isoprenaline stimulation (control: 48.63±1.625%, n=98; vs CPVT: 64.25±1.990%, n=70; Mann-Whitney test; p<0.0001). For hiPSC-SN, CPVT displayed enhanced calcium transients and elevated cAMP elevation following nicotine stimulation, compared to isogenic controls. In hiPSC neurocardiac co-culture, nicotine stimulation increased the beating rate of cardiomyocytes, confirming functional connectivity. The reduction in RR interval was significantly greater in control cardiomyocytes co-cultured with CPVT neurones (n=5) than with control neurones (n=4, Mann-Whitney test; p<0.05). Calcium imaging showed that CPVT hiPSC-SN consistently exhibited elevated calcium transients, regardless of cardiomyocyte genotype they were paired with. This indicates that hyperactivity of neurones is intrinsic and not modulated by co-cultured cardiomyocytes.
Conclusion: Our investigation confirms hallmark CPVT abnormalities in hiPSC-CMs, including arrhythmia, calcium dysregulation and altered β-adrenergic cAMP signalling. Additionally, our results suggest a novel pathogenic role for CPVT sympathetic neurones, whose intrinsic hyperactivity exacerbates the cardiomyocyte phenotype via neurocardiac cross-talk. These findings provide new insights into the pathophysiology of CPVT and highlight the potential of targeting sympathetic excitability as a therapeutic strategy for personalised treatments.