
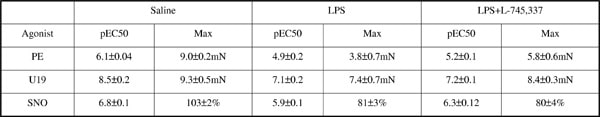
Lipopolysaccharide (LPS)-induced upregulation of endothelial nitric oxide synthase (eNOS) activity is obligatory in the pathogenic expression/function of inducible NOS (iNOS) in sepsis(1). Indeed, LPS-induced iNOS expression, NO synthesis and aortic dysfunction are profoundly depressed in eNOS knockout (KO) mice (2). In contrast we have previously shown that vascular dysfunction in resistance arteries of eNOSKO mice is substantially enhanced and associated with increased plasma cyclooxygenase (COX) metabolites(3). Therefore, we investigated the effect of the COX-2 selective and non-selective inhibitors, L-745,337 and indomethacin (indo) respectively, on LPS-induced vascular dysfunction in eNOSKO mice. Male (25-30g) eNOSKO mice were treated with indo or L-745,337 (5mg/kg, i.p.) 15min prior to saline or LPS (12.5mg/kg, i.v., 4h). Blood samples were collected by terminal cardiac puncture under halothane anaesthesia for 6-keto-PGF1α, thromboxane (TX)B2 and PGE2 measurements using ELISA. Mesenteries were removed for Western blotting for COX-1/2 measurement or 3rd order arteries dissected and mounted in tension myographs. Following normalisation concentration-response curves to the TXA2 mimetic, U-46619 (U19; 0.001-3μM), or phenylephrine (PE; 0.001-30μM) were constructed. Alternatively arteries were precontracted with U19 (~EC80) and relaxation response curves to the NO donor spermine-NONOate (SNO; 0.001-3μM) determined. Whilst COX-2 expression was barely detectable in saline-treated mice profound expression was equally evident following LPS treatment in mesenteries of wild type and eNOSKO mice (n≥3), COX-1 was unaffected (n≥5). Responses to PE, U19 and SNO were suppressed by LPS (n=5-6, p<0.001, table 1); an effect significantly attenuated by L-745,337 (n=5-6, p<0.05), whilst indomethacin had no significant effect (n=4-5). LPS-induced vascular hyporesponsivess was associated with an elevation of plasma 6-keto-PGF1α(≈5 fold),TXB2(≈3 fold) and PGE2(≈34 fold) levels (n=7-9). These were abolished with indo treatment; however, L-745,337 suppressed TXB2(≈4 fold, n=8, p<0.05) and PGE2 (≈14 fold; n=9, p<0.05) only. Together the data suggests a compensatory role for COX-2 derived prostaglandins, in the absence of eNOS, in mediating LPS-induced vascular dysfunction of resistance arteries. COX-2 activity may be important when endothelial dysfunction is prevalent and may explain the poor outcome of NOS inhibitors in the treatment of sepsis.
Life Sciences 2007 (2007) Proc Life Sciences, C61
Research Symposium: Cyclooxygenase-2-derived prostaglandins mediate vascular dysfunction of resistance arteries of endothelial nitric oxide synthase knockout mice in sepsis
S. A. Francis1, A. Ahluwalia1
1. Clinical Pharmacology, William Harvey Research Institute, London, United Kingdom.
View other abstracts by:
Where applicable, experiments conform with Society ethical requirements.