
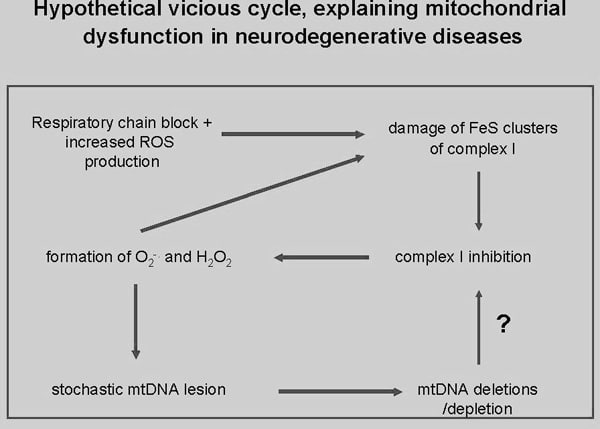
There is compelling evidence for the direct mitochondrial involvement in certain neurodegenerative disorders, like Morbus Parkinson, Friedreichs ataxia, amyotrophic lateral sclerosis, and temporal lobe epilepsy with Ammon’s horn sclerosis. This includes the direct genetic evidence of pathogenic mutations in mitochondrial proteins in certain forms of inherited parkinsonism (like PARK2 – with mutations in parkin, and PARK6 – with mutations in the mitochondrial PTEN-induced kinase 1 (1)) and in Friedreichs ataxia (with mutations in the mitochondrial protein frataxin (2)). Moreover, there is functional evidence for the impairment of mitochondrial respiratory chain in sporadic forms of parkinsonism, amyotrophic lateral sclerosis, and temporal lobe epilepsy with Ammon’s horn sclerosis. In the sporadic forms of the above mentioned neurodegenerative disorders increased oxidative stress appears to be the crucial initiating event which affects respiratory chain function and starts a vicious cycle leading finally to neuronal cell death (cf. Figure). A critical factor which determines the survival of neurons in neurodegenerative disorders is in particular the degree of mitochondrial DNA damage and the maintenance of an appropriate mitochondrial DNA copy number. Clear evidence for a depletion of intact copies of the mitochondrial genome has been provided in all above mentioned neurodegenerative disorders, including amyotrophic lateral sclerosis (3), and temporal lobe epilepsy with Ammon’s horn sclerosis.(4). In the present review we provide a summary and a critical discussion of recently available data.
Life Sciences 2007 (2007) Proc Life Sciences, SA51
Research Symposium: Mitochondrial function in neurodegenerative disorders
M. Baron1, A. P. Kudin1, W. S. Kunz1
1. University of Bonn, Bonn, Germany.
View other abstracts by:
Where applicable, experiments conform with Society ethical requirements.