
Tumour necrosis factor alpha (TNF-α) and interleukin 1β (IL-1β), have been implicated in Ca2+ dysregulation and depressed contractility in ventricular tissue (Duncan et al., 2007). These cytokines stimulate several signalling pathways, one of which results in activation of IκB kinases, phosphorylation of IκB and subsequent translocation of nuclear factor-κB (NF-κB) to the nucleus to modulate gene transcription. Inhibition of NF-κB translocation with IMD-0354 (IC50 ~250 nM) during ischaemia/reperfusion injury reduces infarct size and improves functional recovery (Onai et al., 2004). Our aim was to investigate the role of NF-κB signalling in altered Ca2+ regulation induced by TNF-α and IL-1β in ventricular myocytes. Rats (~250g) were sacrificed humanely and ventricular myocytes prepared using a standard collagenase/protease dispersion technique. Cells were loaded with fura-2 AM, superfused with Tyrode solution (30 °C) and stimulated electrically (1 Hz). Data are mean ± SEM and statistical comparisons made with paired or unpaired t-tests as appropriate or Friedman Repeated Measures ANOVA on ranks. In contrast to Onai et al. (2004), our initial experiments showed IMD-0354 to be toxic; exposure to 0.1 µM IMD-0354 led to an initial increase in contractility (by 60 ± 13%, n=11; P<0.001) followed by failure of both contractions and Ca2+ transients (Fig. 1A) before the cell entered rigor, reminiscent of complete metabolic inhibition (Lancaster & Harrison, 1998). Time from application to rigor was dose-dependent; 335±28, 417±93, 499±32, 751±78 s at 1.0, 0.3, 0.1 and 0.03 μM IMD-0354, respectively. The initial increase in contractility was associated with an increase in myofilament sensitivity (P<0.05, n=11) assessed from plots of cell length vs fura-2 fluorescence (Spurgeon et al., 1992). During the phase of contractile failure myofilament sensitivity was significantly decreased (P<0.05, n=11), time to peak contraction accelerated (P=0.002), time to half relaxation reduced (P=0.029) whereas time to half decay of the Ca2+ transient was increased (P=0.005, Fig. 1B), consistent with an intracellular acidosis. DMEM and Tyrode supplemented with foetal calf serum (FCS) protected cells from IMD-0354 toxicity (contractility was stable after >38 min exposure to 0.1 µM IMD-0354 with FCS present). Time to the development of a rigor contracture was not delayed significantly by cyclosporin A (P=0.214). These data illustrate that IMD-0354 is toxic at concentrations 10-fold below the IC50 for IκB kinase inhibition unless superfusion solutions contained FCS. This may suggest that IMD-0354 toxicity does not result from blockade of NF-κB activity.
University College Dublin (2009) Proc Physiol Soc 15, C73
Oral Communications: Mechanisms underlying cell toxicity of IMD-0354, an inhibitor of IκB kinase, in isolated rat ventricular myocytes
A. Neep1, M. Hardy1, K. White1, L. Hull1, S. Harrison1
1. Multidisciplinary Cardiovascular Research Centre, Institute of Membrane and Systems Biology, University of Leeds, Leeds, United Kingdom.
View other abstracts by:
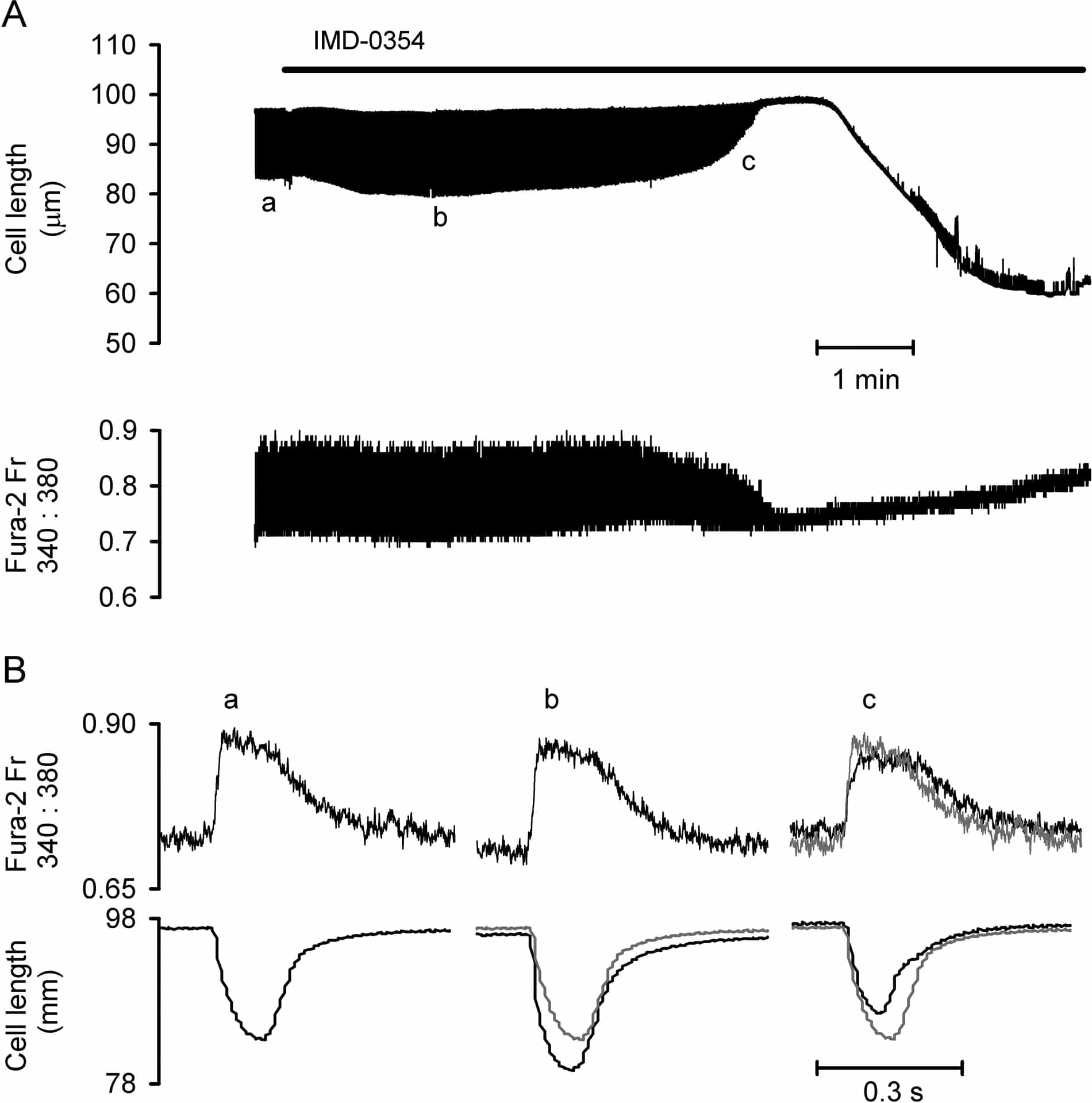
Figure 1. A, cell length and fura-2 fluorescence ratio (Fr) during exposure to 0.1 μM IMD-0354. B, fast time base traces from points a, b and c in panel A. Grey lines are control traces from a for comparison.
Where applicable, experiments conform with Society ethical requirements.