
H441 human airway epithelial cells express an ENaC-like Na+conductance (GNa) that is activated by glucocorticoid hormones (Clunes et al. 2004). Such hormones appear to control ENaC via mechanisms dependent upon serum and glucocorticoid-inducible protein kinase 1 (SGK1), a regulatory kinase that regulates the surface expression of ENaC. Since, the catalytic activity of SGK1 requires the PI3K-regulated phosphylation of SGK1-Ser422/Thr256 (Kobayashi & Cohen, 1999) the present study has explored the effects of a PI3K inhibitor (PI103) upon the glucocortiocoid-evoked increase in GNa seen in these cells. Analysis of currents recorded (Clunes et al. 2004) from glucocorticoid-deprived cells (n = 11) revealed hyperpolarized values of Vm (-47.1 ± 3.9 mV, mean ± s.e.m.) that were unaffected by 10 µM amiloride (-50.4 ± 3.4 mV), whilst dexamethasone-stimulated cells (0.2 µM, 18 – 24 h, n = 9) were depolarized (-7.5 ± 4.8 mV) and consistently displayed hyperpolarizing responses to amiloride (ΔVm = -39.3 ± 5.4 mV, P < 0.001, Student’s paired t test). Further analysis of these data revealed a glucocorticoid-induced, amiloride-sensitive current (IAmil) that reversed at potential (55.7 ± 5.9 mV) close to ENa indicating a high degree of Na+ selectivity. PI103 (0.5 μM, 2 – 3 h) reversed the dexamethasone-induced depolarization of Vm (Control: -0.4 ± 8.1 mV, n = 5; PI103: -51.0 ± 6.0 mV, n = 6; P < 0.001, Student’s t test) and abolished the glucocorticoid-induced Na+ current (Figure 1) and separate studies showed (Western analysis extracted proteins using phospho-specific antibodies) that PI103 also caused complete dephosphorylation of a residue within an endogenous protein (PKB-Ser473) that is phosphorylated in a PI3K-dependent manner (Bayascas & Alessi, 2005). It is therefore clear that PI103 causes complete inactivation of PI3K under the present conditions, and that the dexamethasone-induced increase in GNa is dependent upon PI3K. However, despite this clear finding, dexamethasone (0.2 μM, 0 – 6 h) did not alter PKB-Ser473 phosphorylation indicating that glucocorticoids do not activate PI3K (n = 6). Dexamethasone did, however, induce phosphorylation of an endogenous SGK1 substrate (NDRG1-Thr346/356/366, see Inglis et al. 2009) and this response peaked after 2 – 3 h, and was abolished by PI103 (n = 5). Whilst PI3K is critically important to the glucocorticoid-induced increase in IAmil, the present data indicate that this kinase acts permissively. Dexamathasone may thus induce de novo synthesis of SGK1 whilst basal PI3K activity confers catalytic activity upon the newly sythesised protein by phosphorylating SGK1-Ser422/Thr256(Kobayashi and Cohen, 1999).
University of Manchester (2010) Proc Physiol Soc 19, PC141
Poster Communications: A permisive role for phosphoinositide-3-kinase (PI3K) in the glucocorticoid-induced activation of epithelial Na+ channels (ENaC) in H441 human airway epithelial cells
G. Watt1, S. C. Land1, S. M. Wilson1
1. Centre for Cardiovascular and Lung Reasearch, University of Dundee, Dundee, United Kingdom.
View other abstracts by:
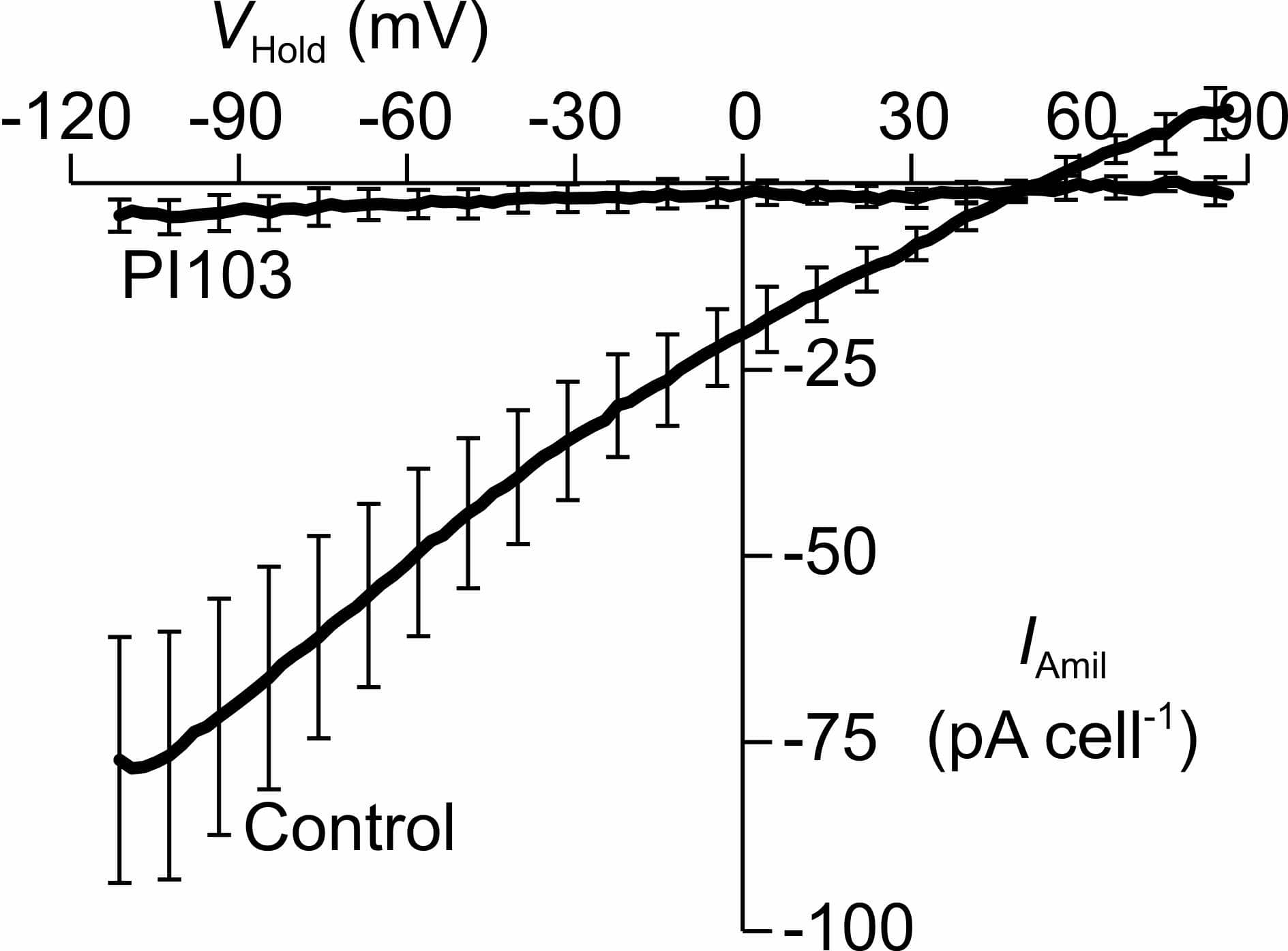
Membrane currents evoked by ramp changes in Vm were quantified under quasi-physiological ionic conditions (see Clunes et al. 2004), firstly under standard conditions and then in the presence of 10 μM amiloride. The current that persisted in the presence of amiloride was then subtracted from the current recorded under standard conditions to isolate the amiloride-sensitive component of the whole cell current (IAmil) which is plotted (mean ± s.e.m) against VHold. All data were recorded from dexamethasone-treated (0.2 µM, 18 - 24 h) cells that had either maintained under control condition (n = 5) or exposed to 0.5 µM PI103 for 2 - 3 h. The magnitudes of all recorded currents have been normalised to the mean input capcitance associated with a single cell (35 pF) in to ensure that variations between the sizes of different cells do not contribute to the variability in the presented data.
Where applicable, experiments conform with Society ethical requirements.