
Physiology News Magazine

NOS1, S-nitrosylation and cardiac contraction
Nitric oxide (NO) is a major regulator of cardiac contraction in both healthy and diseased hearts. NO is produced via enzymes termed nitric oxide synthase (NOS). The neuronal NOS (NOS1) isoform alters key cardiac proteins via post-translational modifications resulting in enhanced contractile function. Therefore, it provides a potential therapeutic target for clinic treatment and new drug development.
Features
NOS1, S-nitrosylation and cardiac contraction
Nitric oxide (NO) is a major regulator of cardiac contraction in both healthy and diseased hearts. NO is produced via enzymes termed nitric oxide synthase (NOS). The neuronal NOS (NOS1) isoform alters key cardiac proteins via post-translational modifications resulting in enhanced contractile function. Therefore, it provides a potential therapeutic target for clinic treatment and new drug development.
Features
Honglan Wang and Mark T. Ziolo
Department of Physiology and Cell Biology, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA
https://doi.org/10.36866/pn.82.36

Contraction of cardiac myocytes occurs by a process termed excitation–contraction coupling (ECC) (Bers, 2002). The initiating event is the opening of L-type Ca2+ channels (ICa), which leads to a release of Ca2+ from the sarcoplasmic reticulum (SR) via the SR Ca2+ release channel (ryanodine receptor, RyR2) (i.e. Ca2+ transient). Upon RyR2 opening, the large efflux of Ca2+ will diffuse and then activate the myofilaments to contract. During relaxation, the majority of Ca2+ is resequestered into the SR by the SR Ca2+-ATPase–phospholamban (SERCA–PLB) complex while the remaining Ca2+ is removed from the cell by the Na+–Ca2+ exchanger.
Nitric oxide and cardiac function
Within cardiac myoctes, nitric oxide (NO) is endogenously produced via two constitutively expressed enzymes: endothelial NO synthase (NOS3) and neuronal NO synthase (NOS1). Another isoform (inducible NO synthase, NOS2) is not constitutively expressed, but its expression is induced during an inflammatory response. NO has been shown to play an important role in both physiological and pathological states. Depending on its source of production, NO may be beneficial or detrimental to cardiac function and remodelling. Although NO is a highly diffusible gas, signalling via the two constitutively expressed isozymes (NOS1 and NOS3) is compartmentalized and each isoform modulates cardiac function differently (Barouch et al. 2002; Ziolo et al. 2008).
NOS3 is localized to the plasmalemmal caveolae. It has been shown that NOS3 signalling decreases the β-adrenergic receptor (AR)-stimulated ICa via activation of the cGMP-dependent protein kinase. NOS3 signalling ultimately decreases the functional response to β-AR stimulation and protects against arrhythmias (Champion et al. 2004; Massion et al. 2004; Wang et al. 2008b).
Unlike NOS3, NOS1 is localized to the SR along with RyR2. NOS1 signalling in the myocyte leads to enhanced contraction and accelerated relaxation. Another difference compared to NOS3 is that NOS1 targets RyR2 (Gonzalez et al. 2007; Wang et al. 2010) and PLB (Wang et al. 2008a) through a cGMP-independent pathway.
RyR2 S-nitrosylation and activity in NOS1 knockout hearts
S-nitrosylation has emerged as an important protein post-translational modification related to cellular signal transduction (Ziolo, 2008). RyR2 contains sulfhydryl residues which can be modified by NO through S-nitrosylation (Xu et al. 1998). Our recent study demonstrated that there were decreased RyR2 S-nitrosylation levels in NOS1 knockout (NOS1–/–) hearts (Wang et al. 2010) (Fig. 1). Along with the deceased RyR2 S-nitrosylation levels, NOS1–/– myocytes also displayed reduced RyR2 activity. We employed various techniques to measure RyR2 activity ranging from direct single RyR2 protein activity to indirect measurements in intact cardiac myocytes.
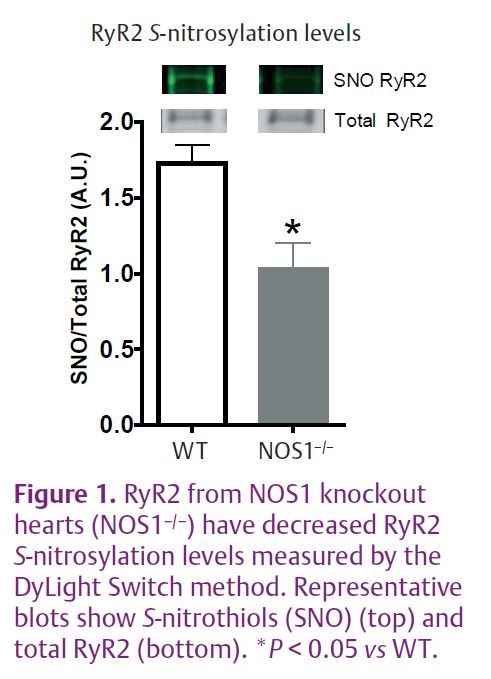
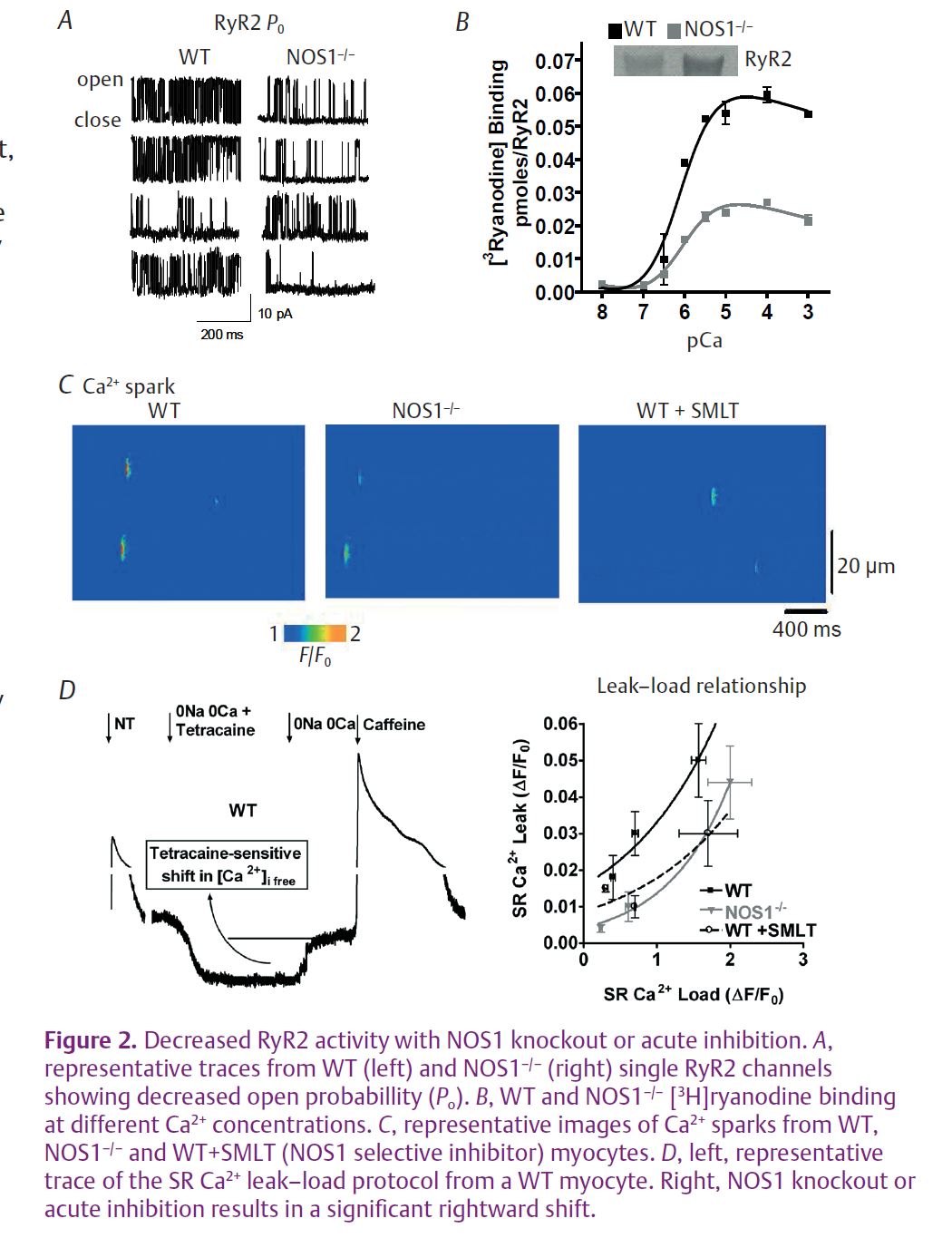
Data from single RyR2 channel recordings showed that the knockout of NOS1 resulted in a 3-fold reduction in RyR2 channel open probability (Po) compared to RyR2 channels from wild-type (WT) hearts (Fig. 2A). In the physiological Ca2+ range (pCa 7 to pCa 3), NOS1 knockout resulted in decreased [3H] ryanodine binding to RyR2 (Fig. 2B) in SR vesicle preparations, indicative of decreased Po. In intact mouse myocytes, NOS1 knockout or acute inhibition significantly reduced Ca2+ spark frequency and amplitude (Fig. 2C). Finally, in a more physiologically relevant approach, NOS1–/– myocytes and WT myocytes with acute NOS1 inhibition (SMLT) had a significant rightward shift in the SR Ca2+ leak–load relationship compared to control WT myocytes (Fig. 2D). Thus, in four distinct methods to measure RyR2 activity, we consistently found impaired RyR2 activity when NOS1 was absent or acutely inhibited.
NOS1–/– hearts and myocytes exhibit a blunted force–frequency response and decreased contraction (Barouch et al. 2002; Wang et al. 2008a).
We suggest that decreased RyR2 activity via reduced S-nitrosylation levels observed in NOS1–/– hearts contribute to the depressed contraction. If this concept is correct, then addition of a NO donor (or
S-nitrosylating agent) should be able to reverse the reduced RyR2 activity by increasing S-nitrosylation leading to increased myocyte contraction.
Can a NO donor increase NOS1–/– RyR2 activity?
SNAP (S-nitroso-N-acetyl-DL-penicillamine), a NO donor and S-nitrosylating agent, was used to test our hypothesis. In NOS1–/–mouse hearts, SNAP was able to increase RyR2 activity in our distinctive methods. That is, SNAP significantly increased RyR2 Po, enhanced both Ca2+ spark frequency and amplitude, and caused a leftward shift in the SR Ca2+ leak–load relationship. There was little effect of SNAP in WT hearts. More importantly, SNAP now normalized RyR2 activity in the NOS1–/– hearts to WT levels. Taken together, SNAP is able to reverse the blunted RyR2 activity.
Can the increased RyR2 activity by SNAP reverse the depressed contraction observed in NOS1–/– myocytes?
Contraction in myocytes was determined by simultaneous measurement of Ca2+ transients and cell shortening. SNAP resulted in an increase in Ca2+ transient and cell shortening amplitudes in WT myocytes, NOS1–/– myocytes and WT myocytes with acute NOS1 inhibition (Fig. 3). However, SNAP had a much larger effect in myocytes with NOS1 knockout or acute inhibition. More importantly, SNAP now normalized the Ca2+ transient and cell shortening amplitudes in these myocytes. Thus, SNAP is indeed able to reverse the depressed contraction in NOS1–/– myocytes.
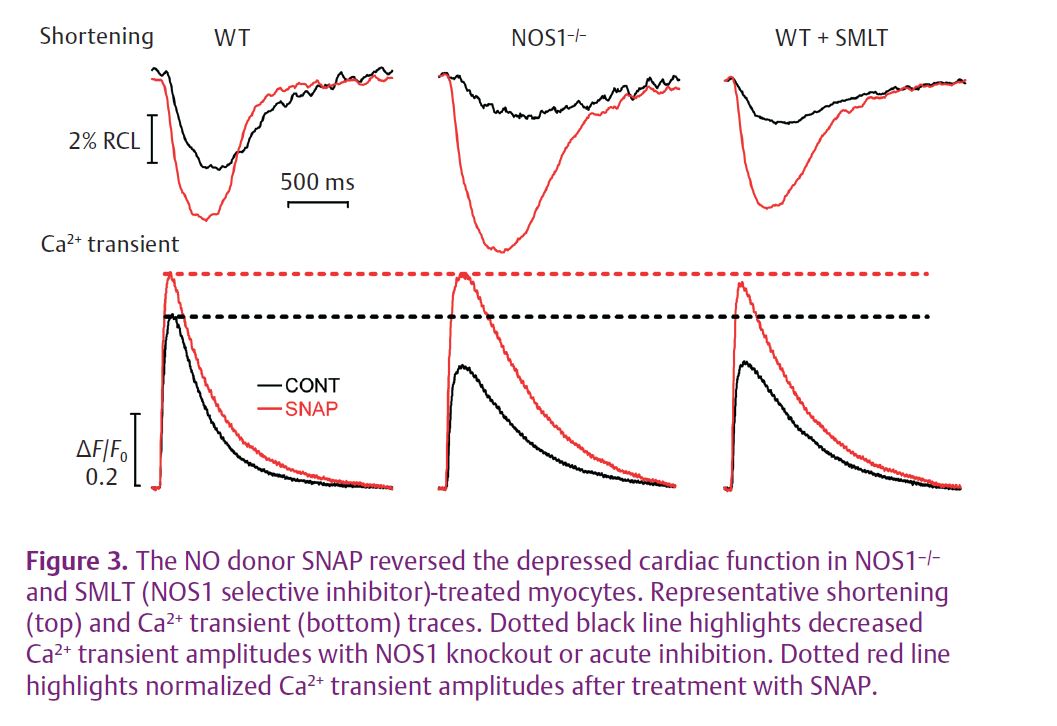
Is RyR2 S-nitrosylation involved in the effects mediated by SNAP?
In NOS1–/– mouse hearts, SNAP increased RyR2 S-nitrosylation levels. As with contraction, SNAP had a much larger effect on NOS1–/– RyR2 S-nitrosylation compared to WT RyR2. Further, in the presence of SNAP, RyR2 S-nitrosylation levels were now normalized. These data suggest that the effect of SNAP to reverse the contraction is, in part, via increased RyR2 S-nitrosylation.
In conclusion, NOS1 signalling results in S-nitrosylation of RyR2, increasing its activity that ultimately leads to enhanced contraction. Therefore, the NOS1-mediated post-translational modifi cation of RyR2 is an important modulator of cardiac myocyte function and may be a useful target for clinical treatment of cardiomyopathies.
References
Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA et al. (2002). Nitric oxide regulates the heart by spatial confi nement of nitric oxide synthase isoforms. Nature 416, 337–339.
Bers DM (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. Champion HC, Georgakopoulos D,
Takimoto E, Isoda T, Wang Y & Kass DA (2004). Modulation of in vivo cardiac function by myocyte-specifi c nitric oxide synthase-3. Circ Res 94, 657–663.
Gonzalez DR, Beigi F, Treuer AV & Hare JM (2007). Defi cient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A 104, 20612–20617.
Massion PB, Dessy C, Desjardins F, Pelat M, Havaux X, Belge C et al. (2004). Cardiomyocyte-restricted overexpression of endothelial nitric oxide synthase (NOS3) attenuates β-adrenergic stimulation and reinforces vagal inhibition of cardiac contraction. Circulation 110, 2666–2672.
Wang H, Kohr MJ, Traynham CJ, Wheeler DG, Janssen PM & Ziolo MT (2008a). Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. Am J Physiol Cell Physiol 294, C1566–C1575.
Wang H, Kohr MJ, Wheeler DG & Ziolo MT (2008b). Endothelial nitric oxide synthase decreases β-adrenergic responsiveness via inhibition of the L-type Ca2+ current. Am J Physiol Heart Circ Physiol 294, H1473–H1480.
Wang H, Viatchenko-Karpinski S, Sun J, Gyorke I, Benkusky NA, Kohr MJ, Valdivia HH, Murphy E, Gyorke S & Ziolo MT (2010). Regulation of myocyte contraction via neuronal nitric oxide synthase: role of ryanodine receptor S-nitrosylation. J Physiol 588, 2905–2917. http://jp.physoc.org/content/588/15/2905.long
Xu L, Eu JP, Meissner G & Stamler JS (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237.
Ziolo MT (2008). The fork in the nitric oxide road: cyclic GMP or nitrosylation? Nitric Oxide 18, 153–156.
Ziolo MT, Kohr MJ & Wang H (2008). Nitric oxide signaling and the regulation of myocardial function. J Mol Cell Cardiol 45, 625–632.