
Physiology News Magazine

Parathyroid hormone-related protein (PTHrP): a modulator of fetal growth and development
The role of PTHrP during embryonic development is slowly being elucidated. Recent data suggest that PTHrP plays a key role in the regulation of placental calcium transport, fetal skeletal development and calcium homeostasis. Our understanding of how PTHrP exerts these effects in utero has been improved by studies using genetically modified mice in which fetal expression of PTHrP has been ablated. These investigations have provided insights into the multiple roles of fetal PTHrP in normal embryonic development
Features
Parathyroid hormone-related protein (PTHrP): a modulator of fetal growth and development
The role of PTHrP during embryonic development is slowly being elucidated. Recent data suggest that PTHrP plays a key role in the regulation of placental calcium transport, fetal skeletal development and calcium homeostasis. Our understanding of how PTHrP exerts these effects in utero has been improved by studies using genetically modified mice in which fetal expression of PTHrP has been ablated. These investigations have provided insights into the multiple roles of fetal PTHrP in normal embryonic development
Features
Mark R Dilworth & Jocelyn D Glazier
Maternal and Fetal Health Research Group, Research School of Clinical and Laboratory Sciences, Manchester Academic Health Science Centre, University of Manchester, St Mary’s Hospital, Manchester, UK
https://doi.org/10.36866/pn.75.22

In order for a baby to undergo normal development and achieve its growth potential, maintenance of an optimal in utero environment is absolutely crucial. Epidemiological evidence has demonstrated that babies of low birth weight, reflecting possible undernourishment during intrauterine life, are at increased risk of various chronic diseases including the development of osteoporosis later in life (Cooper et al. 2002). So, is there is a causal relationship between sub-optimal fetal nutrition and aberrant bone development? Observations from animal studies suggest that there is. Maternal protein restriction leads to reduced bone mineral content and bone area in the offspring, with evidence of altered bone morphology and structural strength (Lanham et al. 2008). Thus, osteoporosis could be partly programmed in utero. To gain a better understanding of the biological mechanisms that link an altered fetal nutrient provision to compromised bone development in utero, we first need to elucidate how fetal skeletal development is regulated, particularly as the placenta must ‘adapt’ its transport function in order to meet the dynamic fetal demand for calcium and mineral provision during skeletogenesis. This demand is most acute over the last third of pregnancy when fetal deposition of calcium rises exponentially. Such an increase in fetal calcium accretion must reflect an increased net calcium flux to the fetus, represented as the difference between the unidirectional maternofetal and fetomaternal fluxes.
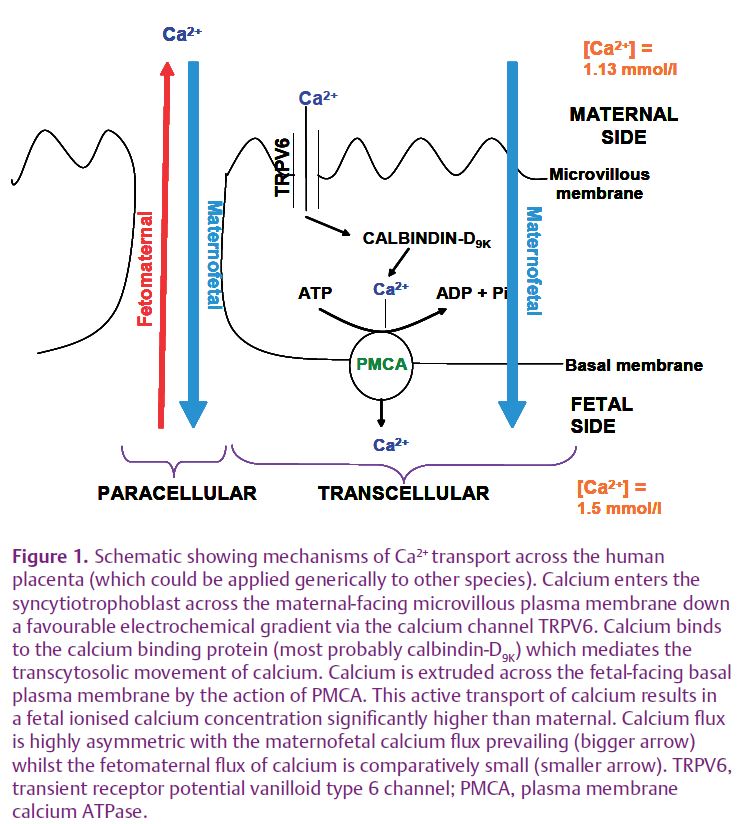
Movement of calcium across the placenta is an asymmetric process, with the maternofetal calcium flux predominant, driven by active, transporter-mediated transfer of calcium. The mechanisms involved in maternofetal calcium flux across the epithelium of human placenta, the syncytiotrophoblast, have been modelled as a three-stage process (Fig. 1): calcium entry, cytosolic translocation and exit, in common with the transcellular movement of calcium across other epithelia such as the intestine and kidney. Ca2+ in maternal plasma is transported across the maternal-facing microvillous plasma membrane of the syncytiotrophoblast down the favourable electrochemical gradient through epithelial calcium channels of the transient receptor potential vanilloid (TRPV) subfamily, with involvement of TRPV6 implicated. Ca2+ is then translocated across the cytosol following binding to calcium binding proteins which help to buffer intracellular Ca2+ whilst facilitating transcellular calcium movement. The molecular identities of the calcium binding proteins involved in this process in human placenta are not well defined, as several calcium binding proteins are expressed. However, in rat placenta and mouse placenta, calbindin-D9K is particularly important as shown by the marked increase in placental expression of calbindin-D9K over the last third of pregnancy and its stoichiometric relationship with the rise in maternofetal calcium flux (Glazier et al. 1992; Bond et al. 2008). In these species, at least, placental calbindin-D9K expression may be rate-limiting to placental calcium transport. Ca2+ is then transported across the fetal-facing basal plasma membrane (BM) of the syncytiotrophoblast against the electrochemical gradient to the fetus. This occurs through the actions of the plasma membrane Ca2+-ATPases (PMCAs) localised to the BM. PMCA1 and PMCA4 isoforms have been demonstrated in placenta (Strid & Powell, 2000; Bond et al. 2008). The upregulation of PMCA activity in BM over the last trimester of human pregnancy (Strid & Powell, 2000) would serve to promote fetal provision of calcium at a time of great fetal demand.
PMCA activity in BM can be stimulated by physiological concentrations of PTHrP (38–94 amide), but not by PTHrP (1–34), PTHrP (67–86) or PTH at comparable concentrations (Strid et al. 2002). Calcium transfer into the fetal circulation of the in situ perfused placenta of parathyroidectomised or thyroparathyroidectomised fetal lambs was also stimulated by the addition of partially purified hPTHrP or recombinant PTHrP(1–84), PTHrP(1–108), PTHrP(1–141), hPTHrP (75–84), hPTHrP(67–86 amide) and hPTHrP(75–86 amide) but not by synthetic PTHrP(1–34) (Abbas et al. 1989; Care et al. 1990). By contrast, infusion of hPTHrP(1–34) or hPTHrP(75–86)amide into the fetoplacental circulation of intact rat fetuses had no effect on placental calcium transport (Shaw et al.1991). These observations, although not consistent between studies, do raise the possibility that PTHrP can act to modulate placental calcium transport and highlight that these effects are PTHrP fragment specific. As PTHrP has the ability to act in an endocrine, paracrine, autocrine as well as intracrine manner, the question arises whether PTHrP produced by the maternal (uterine) and/or fetal (from placenta and fetal tissues) compartments of the uteroplacental unit are involved in such regulation?
We (Bond et al. 2008), and others (Kovacs et al. 1996; Tucci et al. 1996), have explored this issue further by examining fetal calcium homeostasis and placental calcium transport in mice with deletion of the PTHrP gene (PTHrP–/–), whereby the fetal source of PTHrP is eliminated. PTHrP–/– fetuses exhibit chondrodysplasia characterised by the premature and inappropriate ossification of the developing skeleton with skeletal abnormalities apparent, such as a domed skull, short snout and mandible, and shortened upper and lower limbs. One intriguing feature of these animals is that despite the broad tissue distribution of PTHrP, skeletal morphogenesis is most notably affected whilst other tissues show no gross morphological abnormalities (Karaplis et al. 1994). PTHrP–/– fetuses are modestly growth-restricted with an ~6% reduction in fetal weight compared to their wild-type littermates (Kovacs et al. 2001; Bond et al. 2008) and have a significantly reduced blood ionised [Ca2+] resulting in the abolition of the fetomaternal calcium gradient (Kovacs et al. 1996; Tucci et al. 1996; Bond et al. 2008). Near term, they have a significantly higher calcium content than wild-type littermates (Tucci et al. 1996; Bond et al. 2008), implying that the net flux of calcium across the placenta to the fetus has been increased.
To elucidate this further, we measured the unidirectional maternofetal clearance of calcium (CaKmf) across the perfused and intact placenta of PTHrP–/– fetuses compared to wild-type at embryonic day 18 (Bond et al. 2008). These studies demonstrated that CaKmf was significantly raised in PTHrP–/– fetuses whilst calcium flux in the reverse direction, measured as unidirectional fetoplacental clearance of calcium, was unaffected. This stimulation of placental calcium transport appears to be underscored by an upregulated placental expression of calbindin-D9K (Bond et al. 2008). Our data differ from the earlier work of others who showed that, when compared to a marker of diffusion (51Cr-EDTA), CaKmf was reduced in PTHrP–/– fetuses, accompanied by a decrease in placental calbindin-D9K expression and unaltered fetal calcium content (Kovacs et al. 1996). The possible reasons for this have been fully debated previously (Bond et al. 2008).
These studies have clearly demonstrated that fetal PTHrP plays a pivotal role in maintaining the hypercalcaemic status of the fetus relative to the mother. The excessive skeletal mineralization and premature calcification/ossification found in PTHrP–/– fetuses raises the possibility of an elevated fetal calcium demand. This appears to stimulate maternofetal calcium transport by the induction of placental calbindin-D9K expression. The stimuli that drive this response are unknown, but may be multifactorial (Bond et al. 2008).
The widespread tissue expression of PTHrP during fetal life strongly implicates PTHrP as a key regulator of developmental processes that influence fetal growth. In the light of the observations described here, the notion that PTHrP serves as a linchpin in the programming of bone development by in utero environment is certainly worthy of further investigation.
References
Abbas SK, Pickard DW, Rodda CP, Heath JA, Hammonds RG, Wood WI, Caple IW, Martin TJ & Care AD (1989). Stimulation of ovine placental calcium transport by purified natural and recombinant parathyroid hormone-related protein (PTHrP) preparations. Q J Exp Physiol 74, 549–552.
Bond H, Dilworth MR, Baker B, Cowley E, Requena Jimenez A, Boyd RD, Husain SM, Ward BS, Sibley CP & Glazier JD (2008). Increased maternofetal calcium flux in parathyroid hormone-related protein-null mice. J Physiol 586, 2015–2025.
Care AD, Abbas SK, Pickard DW, Barri M, Drinkhill M, Findlay JB, White IR & Caple IW (1990). Stimulation of ovine placental transport of calcium and magnesium by mid-molecule fragments of human parathyroid hormone-related protein. Exp Physiol 75, 605–608.
Cooper C, Javaid MK, Taylor P, Walker-Bone K, Dennison E & Arden N (2002). The fetal origins of osteoporotic fracture. Calcif Tissue Int 70, 391–394.
Glazier JD, Atkinson DE, Thornburg KL, Sharpe PT, Edwards D, Boyd RD & Sibley CP (1992). Gestational changes in Ca2+ transport across rat placenta and mRNA for calbindin9K and Ca2+-ATPase. Am J Physiol 263, R930–R935.
Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM & Mulligan RC (1994). Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev 8, 277–289.
Kovacs CS, Chafe LL, Fudge NJ, Friel JK & Manley NR (2001). PTH regulates fetal blood calcium and skeletal mineralization independently of PTHrP. Endocrinology 142, 4983–4993.
Kovacs CS, Lanske B, Hunzelman JL, Guo J, Karaplis AC & Kronenberg HM (1996). Parathyroid hormone-related peptide (PTHrP) regulates fetal-placental calcium transport through a receptor distinct from the PTH/PTHrP receptor. Proc Natl Acad Sci U S A 93, 15233–15238.
Lanham SA, Roberts C, Perry MJ, Cooper C & Oreffo RO (2008). Intrauterine programming of bone. Part 2: alteration of skeletal structure. Osteoporos Int 19, 157–167.
Shaw AJ, Mughal MZ, Maresh MJ & Sibley CP (1991). Effects of two synthetic parathyroid hormone-related protein fragments on maternofetal transfer of calcium and magnesium and release of cyclic AMP by the in-situ perfused rat placenta. J Endocrinol 129, 399–404.
Strid H, Care A, Jansson T & Powell T (2002). Parathyroid hormone-related peptide (38-94) amide stimulates ATP-dependent calcium transport in the basal plasma membrane of the human syncytiotrophoblast. J Endocrinol 175, 517–524.
Strid H & Powell TL (2000). ATP-dependent Ca2+ transport is up-regulated during third trimester in human syncytiotrophoblast basal membranes. Pediatr Res 48, 58–63.
Tucci J, Hammond V, Senior PV, Gibson A & Beck F (1996). The role of fetal parathyroid hormone-related protein in transplacental calcium transport. J Mol Endocrinol 17, 159–164.
Acknowledgements
The research described in this article was supported by a project grant from the Wellcome Trust (Grant number 076026/Z/04/Z). The Maternal and Fetal Health Research Group is supported by the Manchester Academic Health Sciences Centre (MAHSC) and the NIHR Manchester Biomedical Research Centre.