
Physiology News Magazine

Survival by downsizing: N-terminal truncation of cardiac troponin T increases heart efficiency during energetic crisis
An example of molecular evolution that adds modulatory structures in proteins is the N-terminal variable region of troponin T in the regulatory system of striated muscle. Recent studies have demonstrated that the N-terminal segment of cardiac troponin T can be selectively removed by restricted proteolysis during myocardial adaptation to energetic crisis. By reducing the velocity of contraction and elongating the time of ventricular ejection, this mechanism suggests a novel approach to developing treatment for congestive heart failure
Features
Survival by downsizing: N-terminal truncation of cardiac troponin T increases heart efficiency during energetic crisis
An example of molecular evolution that adds modulatory structures in proteins is the N-terminal variable region of troponin T in the regulatory system of striated muscle. Recent studies have demonstrated that the N-terminal segment of cardiac troponin T can be selectively removed by restricted proteolysis during myocardial adaptation to energetic crisis. By reducing the velocity of contraction and elongating the time of ventricular ejection, this mechanism suggests a novel approach to developing treatment for congestive heart failure
Features
J-P Jin & Han-Zhong Feng
Section of Molecular Cardiology, Evanston Northwestern Healthcare and Northwestern University Feinberg School of Medicine, Evanston, IL 60201, USA
https://doi.org/10.36866/pn.74.21

Actin-activated myosin ATPase powers muscle contraction. In skeletal and cardiac muscles, contraction is regulated by Ca2+ via the thin filament-associated troponin–tropomyosin system (Gordon et al. 2000). The troponin complex consists of three protein subunits: the Ca2+-binding subunit troponin C (TnC), the inhibitory subunit troponin I (TnI) and the tropomyosin-binding anchoring subunit troponin T (TnT). Three homologous TnT isoforms exist in fast skeletal, slow skeletal and cardiac muscles. These TnT isoforms have conserved middle and C-terminal regions but a highly variable N-terminal region (Jin et al. 2008). Biochemical studies have identified that the C-terminal region of TnT binds tropomyosin and interacts with TnI, TnC and F-actin, and the middle region of TnT also binds tropomyosin (Perry, 1998). The N-terminal region of TnT, by contrast, does not bind any known proteins in the muscle thin filament (Fig. 1).
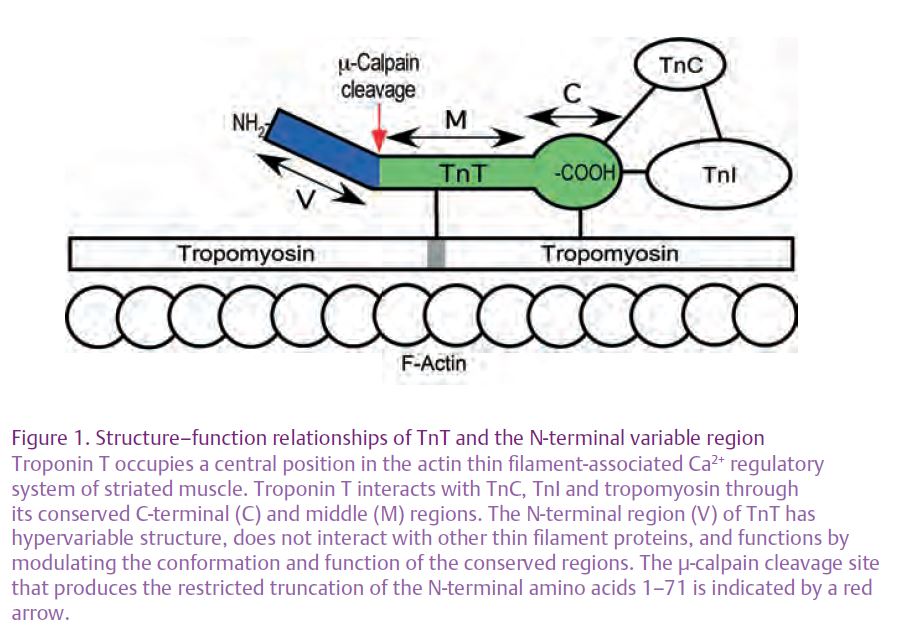
Phylogenetic data have shown that the N-terminal variable region evolved as an addition to the conserved structure of TnT and its deletion does not abolish the Ca2+-dependent activation of actomyosin ATPase in vitro and cardiac muscle contraction in trans-genic mice. Thus, the N-terminal variable region is considered non-essential for baseline TnT function; however, its presence increases actomyosin ATPase activity and sensitivity to Ca2+ activation in reconstituted myofila-ments. Though it does not bind other myofilament proteins, the N-terminal variable region of TnT alters the molecular conformation of the conserved regions, and thereby modulates TnT interaction with TnC, TnI and tropomyosin. Studies of intact TnT isoforms differing in their N-terminal variable regions have demonstrated these functional differences (Jin et al. 2008).
This functional significance of the N-terminal variable region is supported by its regulation by alternative RNA splicing. A cardiac TnT isoform switch occurs during avian and mammalian heart development, which corresponds to a transition from a more acidic, high-molecular weight isoform to a less acidic, low-molecular weight isoform. The molecular mechanism for this isoform switch is the exclusion of exon 5 that encodes 10 mainly acidic residues in the N-terminal region from cardiac TnT (Jin et al. 2008). N-terminal alternative splicing is not limited to cardiac TnT. By inclusion or exclusion of multiple N-terminal coding exons, fast skeletal muscle TnT also undergoes an isoform switch during development (Jin et al. 2008). In addition, slow skeletal muscle TnT exhibits an up-regulation of the low-molecular weight isoform excluding exon 5 in functional adaptation to certain pathological conditions such as type 1 Charcot-Marie-Tooth disease (CMT), an inherited peripheral polyneuropathy (Larsson et al. 2008).
In contrast to transcriptional regulation and RNA splicing, proteolysis is a much more rapid mechanism to regulate protein function. For cardiac TnT, we recently discovered that restricted proteolytic truncation of the N-terminal segment occurs during myocardial ischemia–reperfusion (Zhang et al. 2006). This truncation of TnT is mediated by µ-calpain cleavage and is a selective removal of the N-terminal variable region (Fig. 1), unlike the caspase-mediated TnT proteolysis which removes the N-terminal variable region together with part of the conserved structure, resulting in significant loss of function (Communal et al. 2002). In other words, the calpain-mediated N-terminal truncation of cardiac TnT is regulatory rather than destructive.
Follow up studies confirmed that myocardial infarction in ex vivo rat working hearts via ligation of the left anterior descending coronary artery that supplies the anterior wall of the left ventricle resulted in production of the N-terminal truncated cardiac TnT, not only in the infarct but also in remote areas including the right ventricular free wall. This finding suggests an acute whole-organ proteolytic response triggered by regional ischaemia–reperfusion in the absence of systemic neurohumoral signalling. Left ventricular pressure overload in ex vivo mouse working hearts produced N-terminal truncated cardiac TnT in both ventricles, further suggesting a role of haemodynamic stress in triggering an acute whole-organ proteolytic regulation (Feng et al. 2008).
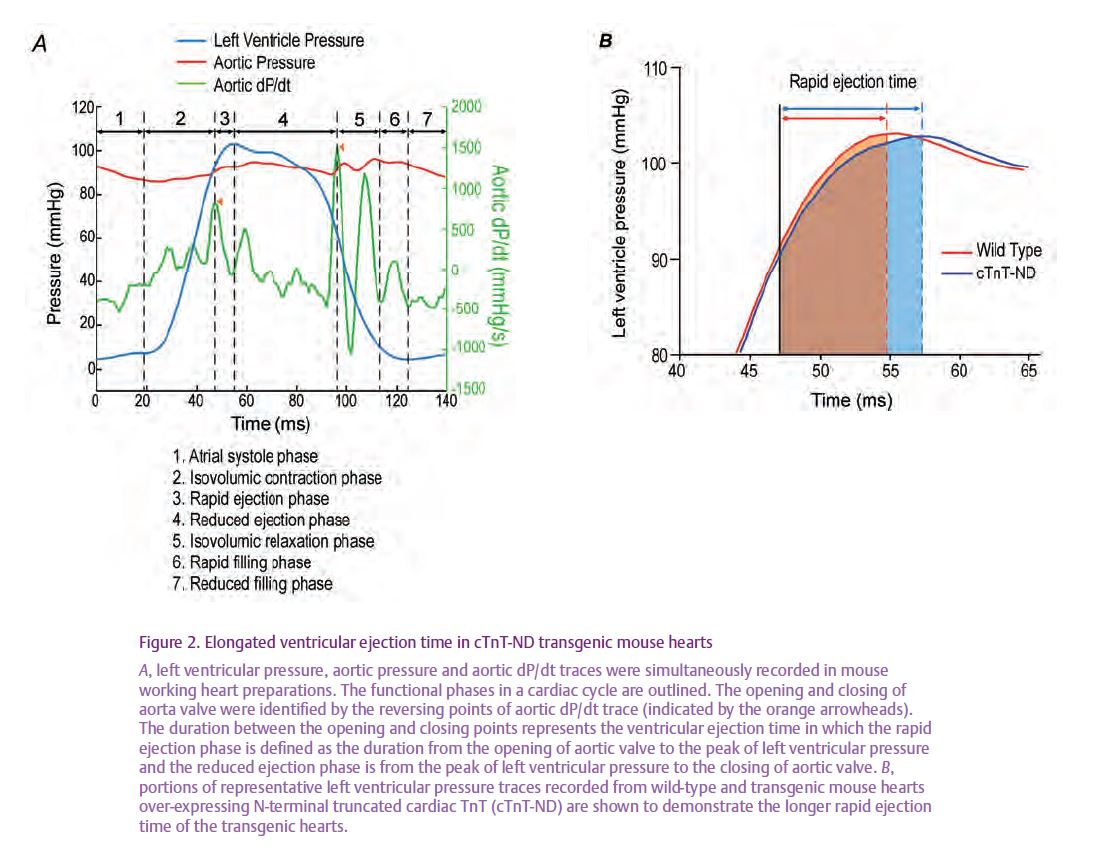
To investigate the pathophysi-ological significance of the N-terminal truncation of cardiac TnT under energetic constraint or workload increase, transgenic mouse hearts were created in which the endogenous intact cardiac TnT was partially replaced by N-terminal truncated cardiac TnT. Functional studies showed that the transgenic mouse hearts had decreased contractile velocity and conse-quently an elongated rapid ejection phase of the cardiac cycle (Fig. 2). Relaxation velocity and ventricular filling time were not affected. These changes would translate into a less powerful heart but also a less energy-consuming pump that is beneficial during energetic crisis. Indeed, when afterload increased, stroke volume decreased in the wild-type but not in the transgenic mouse hearts. The better pumping efficiency of the transgenic mouse hearts demonstrates a novel role of N-terminal truncated cardiac TnT in myocardial adaptation to energetic crisis (Feng et al. 2008). Although the decreased contractile velocity due to N-terminal truncated cardiac TnT could contribute to the depressed myocardial function in ischaemia–reperfusion, it may provide a protection against Ca2+ overload-induced contractures.
Troponin subunits have a rapid turnover rate within cardiac myocytes with a half-life of approxi-mately 3-4 days; therefore, it is plausible that N-terminal truncated cardiac TnT provides a short-term solution for acute ischaemia and other energetic crisis and is replaced with intact cardiac TnT afterwards. In other words, cardiac TnT truncation at the N-terminus seems to switch the cardiac muscle into ‘energy-saving’ mode to sustain heart function during an acute energetic crisis, which may play a role in cardiac protection.
Switching from the faster α-myosin to the slower β-myosin in mouse cardiac muscle increased energy efficiency of the heart (Hoyer et al. 2007). Expression of slower myosin isoforms also increased the energy efficiency of skeletal muscle contraction (Reggiani et al. 1997). The finding that the restricted N-terminal truncation of cardiac TnT resulted in slower contractile velocity demonstrated that modification of TnT structure and function could alter myosin cross-bridge kinetics and affect muscle energetic efficiency. The mechanism for TnT to modulate contractile velocity remains to be investigated. Much more also remains to be learned of the cellular mechanisms that regulate this restricted proteolytic modification of cardiac TnT, and more impor-tantly, how these findings can be used to develop new molecular approaches for the prevention and treatment of myocardial deficiency and congestive heart failure. Heart disease is currently a leading cause of death worldwide, and thus our progress in this study, we hope, will hold immense potential and promise in the years to come.
References
Communal C, Sumandea M, de Tombe P, Narula J, Solaro RJ & Hajjar RJ (2002). Functional consequences of caspase activation in cardiac myocytes. Proc Natl Acad Sci U S A 99, 6252–6256.
Feng HZ, Biesiadecki BJ, Yu ZB, Hossain MM & Jin JP. (2008) Restricted N-terminal truncation of cardiac troponin T: a novel mechanism for functional adaptation to energetic crisis. J Physiol 586, 3537–3550.
Gordon AM, Homsher E & Regnier M (2000). Regulation of contraction in striated muscle (Review). Physiol Rev 80, 853–924.
Hoyer K, Krenz M, Robbins J & Ingwall JS (2007). Shifts in the myosin heavy chain isozymes in the mouse heart result in increased energy efficiency. J Mol Cell Cardiol 42, 214–221.
Jin JP, Zhang Z & Bautista JA (2008). Isoform diversity, regulation and functional adaptations of troponin and calponin (Review). Crit Rev Eukaryot Gene Expr 18, 93–124.
Larsson L, Wang X, Yu F, Höök P, Borg K, Chong SM & Jin JP. (2008) Adaptation by alternative RNA splicing of slow troponin T isoforms in type 1 but not type 2 Charcot-Marie-Tooth disease. Am J Physiol Cell Physiol 295, C722–C731.
Perry SV (1998). Troponin T: genetics, properties and function (Review). J Muscle Res Cell Motil 19, 575–602.
Reggiani C, Potma EJ, Bottinelli R, Canepari M, Pellegrino MA & Stienen GJ (1997). Chemo-mechanical energy transduction in relation to myosin isoform composition in skeletal muscle fibres of the rat. J Physiol 502, 449–460.
Zhang Z, Biesiadecki BJ & Jin JP (2006). Selective deletion of the NH2-terminal variable region of cardiac troponin T in ischemia reperfusion by myofibril-associated mu-calpain cleavage. Biochemistry 45, 11681–11694.
Acknowledgements
The research discussed in this article was supported by grants from the National Institutes of Health (AR048816, HD044824 and HL078773), the National Aeronautic and Space Administration (NNA04Ck26G), the Arnold and Ann Berlin Cardiovascular Care Research Fund, and The Doberman Pinscher Foundation of America to J-PJ.