
Physiology News Magazine

Insight into how the human retina obtains its need of vitamin B1
The retina has a substantial need for a variety of nutrients, including vitamin B1. This vitamin is involved in a variety of metabolic reactions and, thus, is essential for normal functions and well-being of the retina. Hamid Said (below) and colleagues examined how human retinal epithelial cells transport the vitamin, how the transport process is regulated, and what happens when clinical mutations in a thiamin transporter (that occur in the genetic disorder thiamine-responsive megaloblastic anemia) affect the ability of this membrane protein to function as a transporter of the vitamin
Features
Insight into how the human retina obtains its need of vitamin B1
The retina has a substantial need for a variety of nutrients, including vitamin B1. This vitamin is involved in a variety of metabolic reactions and, thus, is essential for normal functions and well-being of the retina. Hamid Said (below) and colleagues examined how human retinal epithelial cells transport the vitamin, how the transport process is regulated, and what happens when clinical mutations in a thiamin transporter (that occur in the genetic disorder thiamine-responsive megaloblastic anemia) affect the ability of this membrane protein to function as a transporter of the vitamin
Features
Hamid M Said
Departments of Medicine and Physiology, University of California School of Medicine, Irvine, CA, USA
https://doi.org/10.36866/pn.70.16

The human retina is a highly differentiated (specialized) tissue whose cells are considered to be among the most metabolically active in the human body. Due to the central role it plays in vision, the retina is protected from the fluctuations in the surrounding environment by what is called the blood-retina barrier (BRB). This barrier also controls access of nutrients, growth factors and hormones to the different cells of the retina. Retinal pigment epithelia (RPE) represent an important part of the BRB and separate the outer retina from its choroidal blood circulation. This single layer of epithelial cells plays many physiological functions that include transport of nutrient/substrates into and from the retina. In order to perform such an important function, cells of the RPE have developed a host of specialized systems for transporting nutrients/ ions/substrates across their plasma membranes. A defect in the ability of these systems will have negative consequences on the well-being of the retinal tissue (Meire et al. 2000; Scharfe et al. 2000). Thus studies into how these systems function, how they are regulated and what intracellular and extracellular factors that affect/regulate their function are of significant importance. Knowledge in these areas has been forthcoming in recent years, especially following the establishment of appropriate cell lines for use in in vitro investigations. Such preparations have eliminated the problems associated with the limited supply of appropriate human RPE cells, their limited survival in culture, the low homogeneity of the primary preparations of these cells, and the tendency of such primary cells to lose their epithelial phenotype with time in culture.
Our recent study examined the mechanism by which vitamin B1 (thiamine) is taken up by cells of the human RPE, determined the molecular identity of the membrane transport systems involved in the process, and identified intracellular and extracellular factors that affect (regulate) the vitamin uptake process (Subramanian et al. 2007). Thiamin is an important micronutrient for the well-being and health of the retina. As with all other cells of the human body, retinal cells cannot synthesize thiamin and thus must obtain the vitamin from the surrounding environment. The vitamin plays an important role as a cofactor in a variety of intracellular metabolic reactions including those involved in energy metabolism. The vitamin also has the ability to reduce cellular oxidative stress and to prevent apoptosis (programmed cell death). Thus, reduction in the level of thiamin in retina cells leads to disturbance in cellular metabolism. Such a reduction in intracellular thiamin level occurs in the genetic disorder of thiamine-responsive megaloblastic anemia (TRMA; also known as Roger’s Syndrome), and leads to retinal abnormalities and visual disturbances in the affected subjects. The cause of TRMA is due to mutations in the human thiamin transporter-1 (hTHTR-1) (Diaz et al. 1999; Fleming et al. 1999; Labay et al. 1999), a major player in moving thiamin into a variety of cell types.
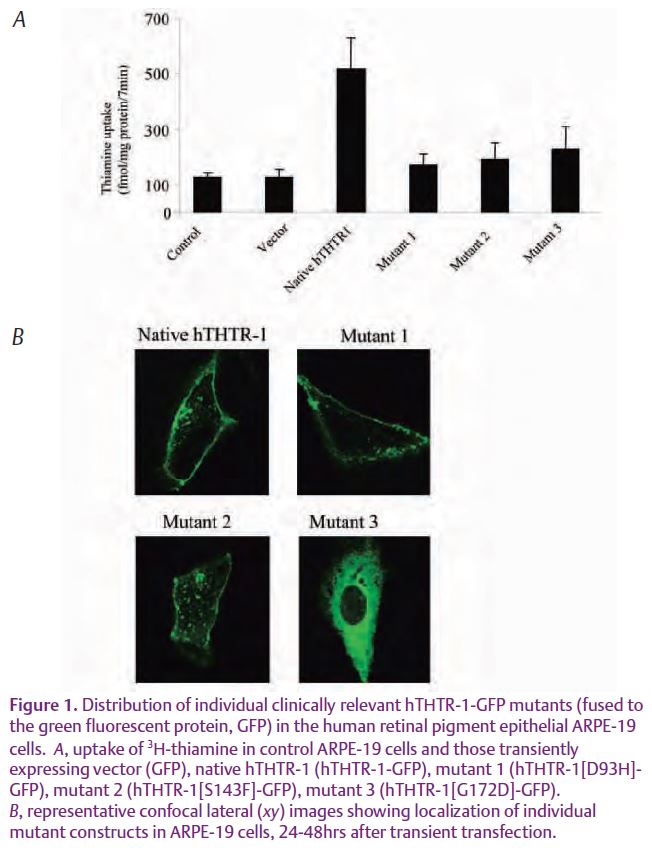
Results of the Subramanian et al studies showed for the first time the existence of an efficient and specific transport process for the internalization of thiamin into cultured human-derived retinal pigment epithelial cells (hRPE). This process was found to be mediated via the function of the two transport systems, the hTHTR-1 and the human thiamin transporter- 2 (hTHTR- 2). These studies also showed that the thiamin uptake by the hRPE cells is adaptively up regulated (turned on) when the cells are maintained in conditions of low thiamin availability via a mechanism that involves activation of genes of both the hTHTR- 1 and hTHTR- 2 (i.e., the SLC19A2 and SLC19A3 genes, respectively). To understand how clinical mutations in the hTHTR-1 that are found in TRMA affect the function of this thiamin transporter and how the cells process the mutant proteins, these investigators generated these mutants experimentally (by means of site-directed mutagenesis) then introduced (transfected) them into cultured hRPE cells. They then followed the movement of the fluorescently labeled proteins inside the living cells by mean of imaging using confocal microscopy. The results showed that these mutations lead to malfunctioning of the hTHTR-1 (Fig. 1A). This was found to be due to either the inability of the cells to move the mutated proteins to the cell membrane or that the cells do move the mutated proteins to the cell membrane, but that the proteins not functional (Fig.1B). Collectively, results of these studies lay the foundation for further investigations into ways to improve the delivery of the vitamin to cells of the human retina, especially in disease conditions associated with low cellular thiamin homeostasis.
References
Diaz GA, Banikazemi M, Oishi K, Desnick RJ & Gelb BD (1999). Mutations in a new gene encoding a thiamine transporter cause thiamine-responsive megaloblastic anaemia syndrome. Nat Genet 22, 309–312.
Fleming JC, Tartaglini E, Steinkamp M, Schorderit DF, Cohen N & Neufeld EJ (1999). The gene mutated in thiamine responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet 22, 305–308.
Labay V, Raz T, Baron D, Mandel H, Williams H, Barrett T, Szargel R, McDonald L, Shalata A, Nosaka K, Gregory S & Cohen N (1999). Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet 22, 300–304.
Meire FM, Van Genderen MM, Lemmens K & Ens-Dokkum MH (2000). Thiamine-responsive megaloblastic anemia syndrome (TRMA) with cone-rod dystrophy. Ophthalmic Genet 21, 243–250.
Scharfe C, Hauschild M, Klopstock T, Janssen AJM, Heidemann PH, Meitinger T & Jaksch M (2000). A noval mutation in the thiamine responsive megaloblastic anaemia gene SLC19A2 in a patient with deficiency of respiratory chain complex I. J Med Genet 37, 669–673.
Subramanian VS, Mohammed ZM, Molina A, Marchant JS, Vaziri Nosratola D & Said Hamid M (2007). Vitamin B1 (thiamine) uptake by human retinal pigment epithelial (ARPE-19) cells: mechanism and regulation. J Physiol 582, 73–85.