
Physiology News Magazine
An integrative approach to control of oxidative phosphorylation during exercise
Features
An integrative approach to control of oxidative phosphorylation during exercise
Features
Brendon J Gurd (1), George J F Heigenhauser (3),Donald H Paterson (1), John M Kowalchuk (1,2)
1: Canadian Centre for Activity and Aging, School of Kinesiology, Faculty of Health Sciences, Department of Physiology and Pharmacology
2: Schulich School of Medicine and Dentistry, The University of Western Ontario, London, Ontario, Canada
3: Department of Medicine, McMaster University, Hamilton, Ontario, Canada L2S 3A1.
https://doi.org/10.36866/pn.69.36
At the onset of exercise there is an instantaneous increase in energy (i.e. ATP) utilization within the active muscle. This increased ATP demand is accompanied by increases in both substrate-level (i.e. anaerobic) and oxidative (i.e. aerobic) phosphorylations, such that muscle ATP content remains relatively constant regardless of the exercise intensity or duration. Muscle O2 consumption, a reflection of mitochondrial oxidative ATP production, can be estimated by pulmonary O2 uptake. During the transition to exercise, pulmonary O2 uptake, and muscle O2 consumption, increase exponentially towards a new level (see Fig. 1, open circles).

As this increase is not ‘instantaneous’, the deficit in meeting ATP requirements by oxidative metabolism must be met by nonoxidative (substrate-level) ATP synthesis. Thus, a consequence of this delay is a metabolic disturbance within the myocyte which includes a breakdown of creatine phosphate
(PCr) and glycogen, and an accumulation of creatine (Cr), ADP, lactate-, protons (H+) and inorganic phosphate (Pi). Together these changes can contribute to increased perception of effort and early fatigue. Therefore, faster activation of muscle O2 consumption and oxidative phosphorylation is expected to reduce cellular disturbances, thereby increasing exercise tolerance and performance.
Control or limitations to the rate of increase of muscle O2 consumption have been linked to; (i) activation of enzymes and provision of substrate (i.e. acetyl CoA and reducing equivalents) to the mitochondrial tricarboxylic acid (TCA) cycle and electron transport chain (ETC); and (ii) adjustments of muscle blood flow and O2 delivery during the transition to exercise. In two recent publications (Gurd et al. 2005, 2006) we demonstrated that the kinetics of pulmonary VO2 (and muscle O2 consumption) during the transition to moderate-intensity exercise (i.e. an intensity domain associated with no, or only a transient, accumulation of lactate-) was faster when preceded by a bout of heavy-intensity, warmup exercise (see Fig. 1, closed circles). Therefore, using this model of prior heavy-intensity exercise, we are attempting to understand the limitations to the activation of muscle O2 consumption and oxidative phosphorylation during the transition to exercise.

Production of ATP by oxidative phosphorylation requires the presence of O2, reducing equivalents (NADH and FADH2 (NADH for simplicity)), ADP and Pi (Fig. 2). While ADP availability, a consequence of an increase in ATP hydrolysis, likely plays a major role in activating mitochondrial oxidative phosphorylation, the availability of other substrates (O2 and NADH) may limit the rate of activation of oxidative phosphorylation at a given ADP concentration ([ADP]) or ratio [ATP]/[ADP][Pi]. A role for the interaction between the concentrations of O2, NADH and ADP in regulating mitochondrial oxidative phosphorylation rather than by a single substrate was reviewed by Wilson (1994). As cellular PO2 decreases below approximately 30 torr, a given rate of mitochondrial respiration can only be sustained by an increase in [NADH] (or [NADH]/[NAD+]) or an increase in [ADP] (or decrease in [ATP]/[ADP]), or both. We suggest that it is this ‘interaction’ between substrate concentrations that both limits kinetics at the onset of exercise, and is responsible for the speeding of pulmonary VO2 kinetics (and muscle O2 consumption) that is observed following a prior bout of heavyintensity, warm-up exercise.
During the transition to exercise, any substrate that is not present in saturating concentrations can limit oxidative phosphorylation. Experimental data obtained from exercising humans demonstrates that intracellular PO2 is approximately 30-40 torr at rest and decreases to less than 10 torr at relatively light-intensities of exercise (Richardson et al. 1995) – i.e. below 30 torr where oxidative phosphorylation begins to display a dependency on intracellular PO2. Technical limitations have prevented an accurate estimation of intramitochondrial [NADH] at rest or during exercise. However, there is evidence that an increase in muscle acetyl group availability via elevated flux through PDH, and subsequently, muscle NADH content can result in a faster activation of oxidative phosphorylation (Howlett et al. 1999). Unlike [O2] and [NADH] which, early in the exercise transition, depend on the adaptation of blood flow and carbohydrate metabolism, respectively, [ADP] begins to increase as soon as muscle contraction is initiated and continues until a new steady-state of ATP synthesis is established. Thus, [ADP] exerts control over the rate of oxidative phosphorylation independent of the concentration of O2 or NADH and can therefore be said to be the driving substrate for oxidative phosphorylation.
Attempts to increase only a single substrate prior to the onset of exercise experimentally have produced varying findings. Studies attempting to increase [O2] have utilized pump perfusion and hyperoxia in animal models, and elevated central (cardiac output) or peripheral blood flow and O2 delivery in humans, most of which failed to speed the rate of pulmonary VO2 (or muscle O2 consumption) (see Tschakovsky & Hughson, 1999 for review). Similarly, attempts to increase [NADH] during the transition to exercise by prior activation of the mitochondrial pyruvate dehydrogenase (PDH) complex and increased flux of carbohydrate-derived substrate into the TCA cycle (by means of dichloroacetate administration) also have yielded mixed results (see Grassi, 2005 for review). However, as discussed earlier it appears that neither O2 nor NADH are present in saturating concentrations and thus should exert control over the rate of oxidative phosphorylation.
We recently demonstrated: i) faster pulmonary VO2 (i.e. oxidative phosphorylation) kinetics following heavy-intensity, warm-up exercise in association with ii) greater muscle perfusion (determined by nearinfrared spectroscopy (NIRS)-derived increase in total and oxygenated haemoglobin-myoglobin concentration) and iii) greater mitochondrial PDH activity which contributes to a greater muscle acetyl CoA and NADH content (determined by muscle biopsy of the quadriceps muscle group) (Gurd et al. 2005; Gurd et al. 2006). The discrepancy between this finding and that of other studies where only a single substrate was augmented (i.e. O2 or NADH, see above) is that following heavy-intensity exercise, both O2 and NADH availability are elevated prior to the onset of exercise and thus are immediately available to the mitochondrial ETC. Therefore, the faster rate of activation of oxidative phosphorylation seen after heavyintensity exercise is a consequence of removing any limitation imposed by either muscle perfusion and O2 delivery, or enzyme activation and NADH availability, or both.
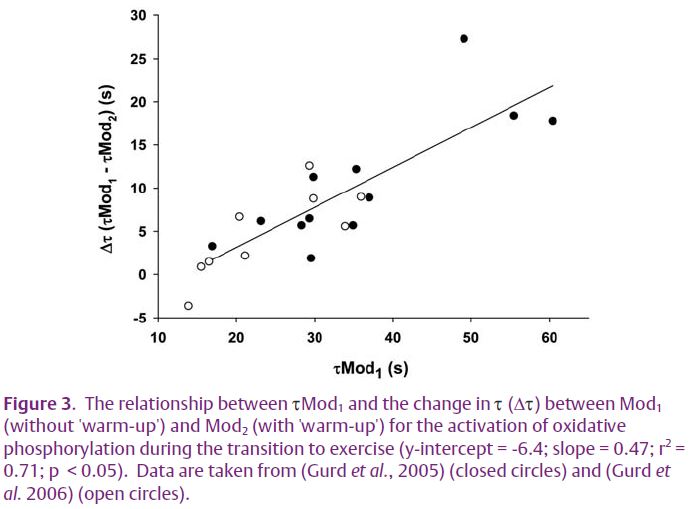
An interesting observation from our recent publications was that the relative speeding of pulmonary VO2 kinetics that was seen in our subjects was not consistent, but was dependent on their initial rate of adaptation (Fig. 3) – i.e. the speeding of kinetics was less in those subjects who presented with faster VO2 kinetics in the ‘no warmup’ condition. Many studies examining control of oxidative phosphorylation have used healthy, physically fit, young adults or animal models using highly capillarized, oxidative muscle preparations. In these instances the adaptation of pulmonary VO2 kinetics and muscle O2 consumption is relatively rapid (time constant (τ) ~ 20 s). Thus, any speeding in the adaptation of oxidative phosphorylation resulting from an intervention in healthy, physically fit, young humans or in oxidative animal muscle preparations is likely to be small and difficult to detect. The situation is likely to be exacerbated in situations where only a single substrate is elevated, such as in the previously mentioned studies where either [O2] or [NADH] was increased alone.
Interestingly, in cases where elevating one or more substrate(s) was successful in speeding the activation of oxidative phosphorylation, full activation was still not instantaneous. This period may represent the time required for ADP to increase to appropriate levels to drive oxidative phosphorylation. Consistent with this idea is the finding that rapid increases in [ADP] alone (either by iodoacetamideinduced inhibition of the creatine kinase (CK) reaction or by using CKknockout mice) result in a significantly faster activation of oxidative phosphorylation during the transition to exercise (for an example see Kindig et al. 2005).
The adaptation of oxidative phosphorylation during the transition to exercise appears to be dependant on the provision of ADP with the availability of O2 and NADH acting as modulators. Our own data show that if, before the start of exercise, muscle perfusion and O2 delivery and muscle substrate (pyruvate) and enzyme (PDH) activities are elevated, the rate of activation of mitochondrial oxidative phosphorylation and O2 consumption (as determined by pulmonary VO2 ) is faster compared to a control condition. As a consequence substrate-level phosphorylations are minimized, which should reduce metabolic disturbances linked to fatigue and exercise intolerance.
References
Grassi B (2005). Delayed metabolic activation of oxidative phosphorylation in skeletal muscle at exercise onset. Med Sci Sports Exerc 37, 1567–1573.
Gurd B, Peters SJ, Heigenhauser GJ, LeBlanc PJ, Doherty TJ, Paterson DH & Kowalchuk JM (2006). Prior heavy exercise elevates pyruvate dehydrogenase activity and speeds O2 uptake kinetics during subsequent moderateintensity exercise in healthy young adults. J Physiol 577, 985–996.
Gurd BJ, Scheuermann BW, Paterson DH & Kowalchuk JM (2005). Prior heavy-intensity exercise speeds VO2 kinetics during moderateintensity exercise in young adults. J Appl Physiol 98, 1371–1378.
Howlett RA, Heigenhauser GJF, Hultman E, Hollidge-Horvet & Spriet LL (1999). Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. Am J Physiol 277, E18–25.
Kindig CA, Howlett RA, Stary CM, Walsh B & Hogan MC (2005). Effects of acute creatine kinase inhibition on metabolism and tension development in isolated single myocytes. J Appl Physiol 98, 541–549.
Richardson RS, Noyszewski EA, Kendrick KF, Leigh JS & Wagner PD (1995). Myoglobin desaturation during exercise. Evidence of limited O2 Transport. J Clin Invest 96, 1916–1926.
Tschakovsky ME & Hughson RL (1999). Interaction of factors determining oxygen uptake at the onset of exercise. J Appl Physiol 86, 1101–1113.
Wilson DF (1994). Factors affecting the rate and energetics of mitochondrial oxidative phosphorylation. Med Sci Sports Exerc 26, 37–43.