
Physiology News Magazine
The amazing versatility of hPEPT1
hPEPT1 is the port of entry into enterocytes of all possible di- and tri-peptides resulting from the digestion of dietary proteins. Since the mid-1990s, it has been exploited as a tool to enhance the oral availability of an ever-increasing number of drugs and prodrugs –both peptidic and non-peptidic. Recently, Monica Sala-Rabanal and colleagues uncovered the molecular mechanism by which peptides and drugs alike make their way into the human enterocytes
Features
The amazing versatility of hPEPT1
hPEPT1 is the port of entry into enterocytes of all possible di- and tri-peptides resulting from the digestion of dietary proteins. Since the mid-1990s, it has been exploited as a tool to enhance the oral availability of an ever-increasing number of drugs and prodrugs –both peptidic and non-peptidic. Recently, Monica Sala-Rabanal and colleagues uncovered the molecular mechanism by which peptides and drugs alike make their way into the human enterocytes
Features
Monica Sala-Rabanal
Department of Physiology, David Geffen School of Medicine at UCLA Los Angeles, CA, USA
https://doi.org/10.36866/pn.66.18

The human proton-coupled oligopeptide cotransporter hPEPT1 (gene SLC15A1) is expressed in the brush border membrane of the enterocytes, the S1 segment of the renal proximal tubules and the hepatic bile ducts. In addition to being responsible for the uptake of all natural di- and tripeptides, this all-purpose transporter has been implicated in the absorption of everyday drugs, such as β-lactam antibiotics. Here, I will highlight the milestones of oligopeptide transport research and review the key molecular, functional and pharmacological aspects of hPEPT1.
The search
The first evidence that intact peptides are transported across the intestinal epithelium was obtained in the late 1950s (Newey & Smyth, 1959). Until the early 1970s, however, the predominant view was that dietary protein was absorbed primarily in the form of free amino acids. This was challenged when it was observed that patients suffering from genetically impaired amino acid transport, as occurs in cystinuria and Hartnup disease, did not develop protein malnutrition. In vivo absorption studies in human volunteers suggested the presence of a low affinity, high capacity transport system for di- and tripeptides, but that would not take longer peptides or single amino acids (see Adibi, 1997). During the 1980s, extensive work in intestinal brush border membrane vesicle (BBMV) preparations and the human colon cancer cell line CaCo-2 confirmed the existence and basic kinetics of the system, and unveiled key features of the mechanisms involved in short-chain peptide absorption. In particular, evidence collected by Leibach and coworkers on rabbit BBMV demonstrated that transport of di- and tripeptides is electrogenic and coupled to an inwardly-directed proton gradient (see Ganapathy & Leibach, 1985). A major breakthrough came with the expression cloning of the first mammalian gene that encoded peptide transport activity, the rabbit PEPT1 (Fei et al. 1994); cloning and functional characterization of the human isoform followed (Liang et al. 1995; Mackenzie et al. 1996b; Sala-Rabanal et al. 2006).
Mechanisms of hPEPT1 function

Fig.1 illustrates the standard model of intestinal peptide absorption. After a meal, proteins are hydrolyzed by pancreatic proteases, and subsequently broken into short-chain peptides by peptidases located in the apical membrane of enterocytes. Di- and tripeptides are then carried into the cytoplasm by hPEPT1, against a concentration gradient and together with protons. Peptides are then released unmodified to the blood –through an unidentified basolateral transporter– or hydrolyzed by the action of endogenous peptidases into amino acids, which then leave the cell via specific transporters. The overall electrochemical balance is maintained by the apical Na+/H+ exchanger NHE-3 and the basolateral Na+/K+-ATPase (Daniel, 2004).
To address the molecular mechanism of apical H+/oligopeptide cotransport, we expressed the cloned hPEPT1 in Xenopus laevis oocytes and performed comprehensive voltage-clamp electrophysiological assays. We found that transport by hPEPT1 is electrogenic, H+–coupled, and voltage dependent (Mackenzie et al. 1996b; Sala-Rabanal et al. 2006). hPEPT1 is capable of transporting neutral and charged substrates; in all cases, transport is associated with the generation of inward cationic currents, regardless of the net charge of the substrate (Mackenzie et al. 1996a).
In addition, hPEPT1 responds to step jumps in membrane potential with transient presteady-state currents or charge movements (Mackenzie et al. 1996a; Sala-Rabanal et al. 2006), that have been postulated to be due to changes in the conformation of the carrier protein as it goes through the transport cycle (Loo et al. 1993).
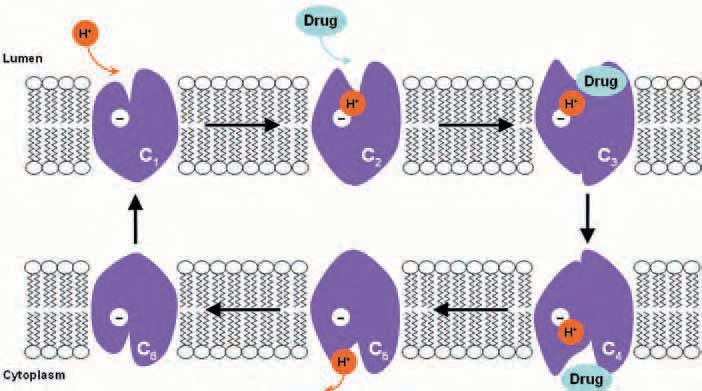
By simultaneous analysis of the kinetics of steady-state dipeptideinduced inward proton currents –to monitor substrate transport– and of presteady-state currents –to monitor the protein conformations during the H+/dipeptide cotransport–, we established a kinetic model for hPEPT1 function (Sala-Rabanal et al. 2006).
The ordered model, summarized in Figure 2, assumes that cotransport happens through a series of conformational changes induced by the binding of ligands (H+ and substrate) and membrane potential. In a transport cycle, one H+ binds to the outsideoriented empty transporter (state C1) to form the proton-carrier complex (C2). Then the substrate binds, and the substrate-loaded protein (C3) undergoes a conformational change (C4) that results in H+/dipeptide cotransport. On the cytoplasmic side, the substrate dissociates (C5), and then the proton is released (C6). A rate-limiting step of the transport cycle is the reorientation of the empty carrier within the membrane (C6 → C1). Variations in dipeptide and drug transport by hPEPT1 are due to differences in affinity and in turnover rate.
Surprising substrate selectivity: peptide bond not a must!
Over the last decade, the substrate specificity of hPEPT1 has been subject to intense scrutiny, driven by the prospect of using the transporter as a way to enhance oral bioavailability of drugs and prodrugs. Recently, comprehensive three-dimensional quantitative structure-activity relationship (3D-QSAR) studies have led to the identification of the minimum structural requisites for hPEPT1 substrates, among which, remarkably, a peptide bond is not included. Thus, the essential features for recognition comprise a simple three-point model: the presence of hydrogen-bond donor and acceptor sites and an electrodense region at given positions within the molecule are sufficient to grant access to the peptide transporter binding site (Biegel et al. 2005).
One direct implication of such unrestrictive steric requirements is that virtually all possible natural di- and tripeptides composed by L-α-amino acids may be transported by hPEPT1 – an impressive 8,400 different substrates. In addition, a large number of pharmacologically active compounds, both peptidomimetic and non-peptidic, satisfy the minimums and have been tested as putative hPEPT1 substrates. The list includes β-lactam antibiotics (penicillins and cephalosporins), inhibitors of the angiotensin-converting enzyme (captopril, enalapril), antineoplastics and prodrugs (valacyclovir).
Drug absorption and structure-affinity studies have been largely based on competition assays (Daniel, 2004; Biegel et al. 2005), mainly because these compounds are not commercially available in radiolabelled form. Competition assays, however, do not allow discrimination between substrates and inhibitors –that is, whether a given drug is transported or simply recognized and bound to the carrier. Using our electrophysiological approach, we demonstrated that βlactams ampicillin, amoxicillin, cephalexin and cefadroxil, and the antineoplastics bestatin and δaminolevulinic acid are indeed transported by hPEPT1, by the same alternating-access mechanism as dipeptides (Fig. 2). These drugs are transported with lower affinity and turnover rate than dipeptides. Our findings implicate that drug absorption by hPEPT1 may be compromised by the presence of physiological concentrations of dietary peptides in the gut. Thus, oral delivery drugs should be taken on an empty stomach (Sala-Rabanal et al. 2006).
Future directions
Despite the unquestionable progress that has been made, there are still gaps in our knowledge about hPEPT1 mediated drug absorption. Given the pharmacological importance of hPEPT1, solving the crystal structure would validate and complement the computational studies and provide a gigantic leap towards the rational design of oral delivery drugs and prodrugs.
Acknowledgements
The author wishes to thank Bruce Hirayama, Donald Loo and Ernest Wright for their advice in the writing of this manuscript.
References
Adibi SA (1997). The oligopeptide transporter (Pept-1) in human intestine: biology and function. Gastroenterology 113, 332-340.
Biegel A, Gebauer S, Hartrodt B, Brandsch M, Neubert K & Thondorf I (2005). Three-dimensional quantitative structure-activity relationship analyses of beta-lactam antibiotics and tripeptides as substrates of the mammalian H+/peptide cotransporter PEPT1. J Med Chem 48, 4410-4419.
Daniel H (2004). Molecular and integrative physiology of intestinal peptide transport. Annu Rev Physiol 66, 361-384.
Fei YJ, Kanai Y, Nussberger S, Ganapathy V, Leibach FH, Romero MF, Singh SK, Boron WF & Hediger MA (1994). Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature 368, 563-566.
Ganapathy V & Leibach FH (1985). Is intestinal peptide transport energized by a proton gradient? Am J Physiol Gastrointest Liver Physiol 249, G153-G160.
Liang R, Fei YJ, Prasad PD, Ramamoorthy S, Han H, Yang-Feng TL, Hediger MA, Ganapathy V & Leibach FH (1995). Human intestinal H+/peptide cotransporter. Cloning, functional expression, and chromosomal localization. J Biol Chem 270, 6456-6463.
Loo DDF, Hazama A, Supplisson S, Turk E & Wright EM (1993). Relaxation kinetics of the Na+/glucose cotransporter. Proc Natl Acad Sci USA 90, 5767-5771.
Mackenzie B, Fei YJ, Getanapathy V & Leibach FH (1996a). The human intestinal H+/oligopeptide cotransporter hPEPT1 transports differently-charged dipeptides with identical electrogenic properties. Biochim Biophys Acta 1284, 125-128.
Mackenzie B, Loo DDF, Fei Y, Liu WJ, Ganapathy V, Leibach FH & Wright EM (1996b). Mechanisms of the human intestinal H+-coupled oligopeptide transporter hPEPT1. J Biol Chem 271, 5430-5437.
Newey H & Smyth DH (1959). The intestinal absorption of some dipeptides. J Physiol 145, 48-56.
Sala-Rabanal M, Loo DDF, Hirayama BA, Turk E & Wright EM (2006). Molecular interactions between dipeptides, drugs and the human intestinal H+ -oligopeptide cotransporter hPEPT1. J Physiol 574, 149-166.