
Physiology News Magazine

Endothelial nitric oxide synthase-derived NO signals regulate microvascular permeability
Regulation of vascular tone is largely dependent on the production of nitric oxide (NO) by the endothelium. Recent findings provide compelling evidence supporting an important role for endothelial NO in the regulation of microvascular permeability
Features
Endothelial nitric oxide synthase-derived NO signals regulate microvascular permeability
Regulation of vascular tone is largely dependent on the production of nitric oxide (NO) by the endothelium. Recent findings provide compelling evidence supporting an important role for endothelial NO in the regulation of microvascular permeability
Features
Walter N Durán, Takuya Hatakeyama*, & Fabiola A Sánchez
Department of Pharmacology & Physiology, New Jersey Medical School, University of Medicine and Dentistry of New Jersey, Newark, NJ, USA and *Department of Surgery, Seisho-kai Memorial Hospital,Tokyo, Japan
https://doi.org/10.36866/pn.65.21

Inflammation is recognized as a fundamental underlying alteration in many diseases. One of the hallmarks of inflammatory processes is an increase in microvascular permeability (hyperpermeability) to macromolecules. An important function of hyperpermeability is to allow the movement of macromolecules for tissue healing and remodelling in response to injurious stimuli. Under inflammatory conditions, microvascular permeability to macromolecules is controlled mainly at the postcapillary venules. The coordination of appropriate physiologic responses to the changing tissue environment in vivo is coordinated through signalling interactions between blood cells, vascular wall and parenchymal cells.
Nitric oxide is recognized as an important signalling regulator of cardiovascular function, but its role in the control of microvascular permeability was regarded as controversial. Evidence in tissues and in isolated venules demonstrated that the activity of endothelial NO synthase (eNOS) increases microvascular permeability to macromolecules in response to inflammatory agents (Ramírez et al. 1995; Yuan et al. 1993). Other results indicated that NOS activity prevents increases in permeability (Kurose et al. 1995). In either view, the evidence for endogenous NO involvement as a positive or negative modulator of permeability was persuasive but not necessarily compelling as it was based mainly on the ability of L-arginine analogs to block NOS non-specifically.
The development of genetically engineered mice has contributed importantly to evaluate more directly the function of eNOS in microvascular permeability, with the appropriate caveats related to the adaptive changes occurring in response to deletion of important genes. Using eNOS-/- mice, we and others demonstrated that deletion of the gene encoding for eNOS prevents mounting an appropriate hyperpermeability response to VEGF (vascular endothelial growth factor) in skin (Fukumura et al. 2001) and to PAF (platelet-activating factor) in cremaster muscle and in mesentery (Hatakeyama et al. 2006). Figure 1 illustrates the dramatic difference in the PAF-induced hyperpermeability response in mouse cremaster. Changes in microvascular permeability were evaluated in the interstitial space by integrated optical intensity (IOI) using fluorescently labelled dextran (FITC-dextran 70, with mean molecular weight = 70,000 daltons) as a macromolecular indicator. Topical application of PAF caused an increment from a baseline value of 2.4 ± 2.2 to a peak net value of 84.4 ± 2.7 units in wild-type mice; whereas in mice lacking the eNOS gene (eNOS-/- mice) PAF increased IOI only from a baseline of 1.0 ± 0.3 to a peak net value of 15.6 ± 7.7 units. Figure 1 also demonstrates the extravasation of FITC-dextran 70 through a series of images captured by intravital microscopy and computer-assisted digital image analysis.
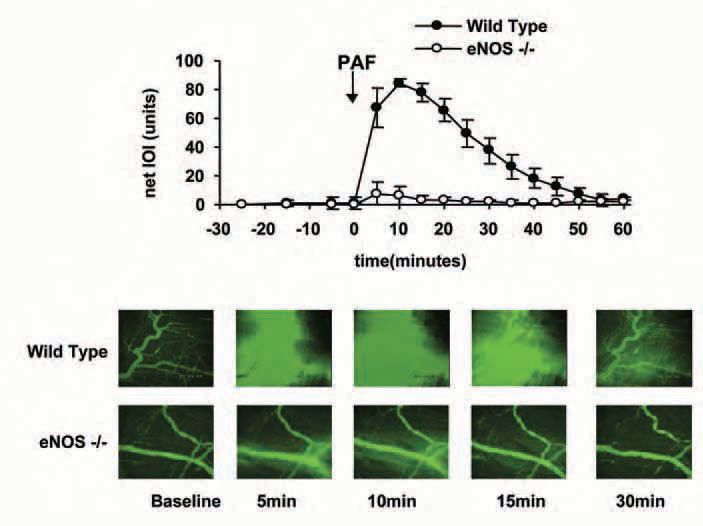
Importantly, we demonstrated that eNOS was the relevant nitric oxide synthase in this NO-regulated response as we established that mice lacking the gene encoding for inducible NOS (iNOS) produced a hyperpermeability to PAF that was of comparable magnitude to that of wild-type mice (Hatakeyama et al. 2006). We also showed that inhibition of eNOS by caveolin-1 scaffolding domain (an endogenous cellular regulator of eNOS activity) reduced the ability of PAF to increase microvascular permeability in cremaster muscle and mesentery of wild-type mice (Hatakeyama et al. 2006). The clinical significance of eNOS-derived NO is manifested by the reduction in postischemic microvascular permeability observed in cremaster muscle of eNOS-/- mice relative to wild-type mice (unpublished data) and by the increase in pulmonary microvascular permeability in caveolin1 deficient mice (Schubert et al. 2002). Several laboratories, in further support of regulation of microvascular permeability by eNOS-derived NO, have documented that inhibition of eNOS decreases transport of macromolecules across monolayers of endothelial cells, whereas substances that phosphorylate eNOS and enhance NO production increase it (Lal et al. 2001; Yuan, 2000).
Mechanisms of signal transduction in hyperpermeability
Much remains to be learned about how eNOS-derived NO signals for permeability regulation in microvascular endothelia. While the gene sequence of eNOS is highly conserved among species, the posttranslational regulatory modalities may vary in different species. A potentially important regulatory mechanism reported early is the phosphorylation of eNOS by Akt (Fulton et al. 1999). However, recent data raise questions as to the specificity conferred to the signal by Akt-induced phosphorylation, particularly at Serine-1179. The questions are based on the observation that, while necessary, this phosphorylation step is common to several different NO-associated vascular functions – such as permeability, vasodilation, angiogenesis, and cell survival (Sánchez et al. 2006). The question is fundamentally valid even though several eNOS phosphorylation sites have been described along with regulation of the enzyme by proteinprotein interactions, some of which may represent novel mechanisms or pathways (Dudzinski et al. 2006).
Enzyme location is of importance to achieve its specific function. Endothelial nitric oxide synthase is found mainly in the cell membrane, but it is also well represented in Golgi under baseline conditions. The relationship between eNOS location and function might be of utmost relevance in endothelium due to the high NO-scavenging capacity of blood haemoglobin. Translocation of eNOS from membrane to other cell compartments in response to agonists has been documented by several techniques including conventional and confocal fluorescence microscopy as well as western blotting. Cellular location of eNOS may represent a mechanism that determines the rate of NO production under different physiological or experimental conditions. Whether translocation represents a defined mechanism or a step in signalling to bring the enzyme closer to its functional target remains to be determined. The diagram in Fig. 2 describes a simplified view of experimental evidence indicating that differential translocation of eNOS is associated to microvascular function. Using agonists that afford a relatively clear discrimination between precapillary and postcapillary functions (basically vasodilation versus permeability), we observed in cells in culture that acetylcholine (a ‘pure’ vasodilator) induces preferential translocation to Golgi, whereas PAF (a hyperpermeability inducing agent) caused translocation preferentially to the cytosol (Sánchez et al. 2006). The basis for the physiological translocation of eNOS in vivo has been demonstrated elegantly in transgenic mice using a fluorescent label that allows tracking the enzyme location in vivo (Cheng et al. 2005).
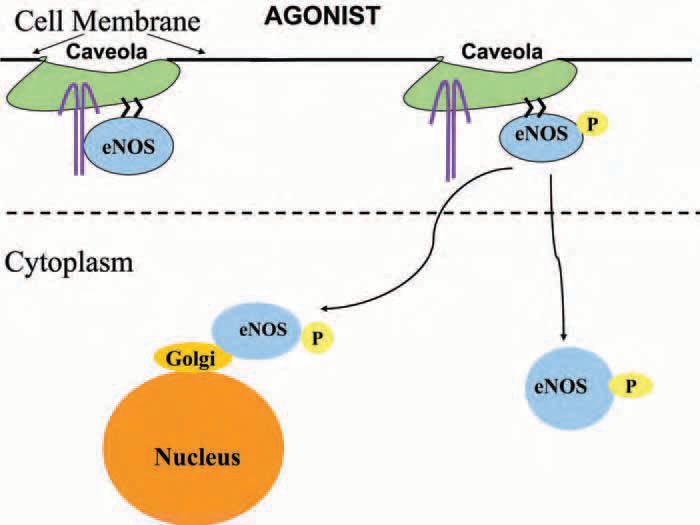
The biological or experimental basis for the earlier controversy regarding NO regulation remains unknown, but it may reside in species differences and in inherent limitations of the use of nonspecific pharmacologic agents. Compelling evidence based on the use of genetically engineered mice demonstrates that eNOS-derived NO regulates the necessary hyperpermeability in postischemic tissues and in inflammation. Despite abundant and sophisticated information obtained in cell cultures, the door is wide open for studies to elucidate the mechanisms by which eNOS-derived NO exactly and specifically regulates microvascular permeability in vivo.
Acknowledgments
Our work was supported by NIH grant 5HL70634.
References
Cheng C, van Haperen R, de Waard M, van Damme LCA, Tempel D, Hanemaaijer L, van Cappellen GWA, Bos J, Slager CJ, Duncker DJ, van der Steen AFW, de Crom R, & Krams R (2005). Shear stress affects the intracellular distribution of eNOS: direct demonstration by a novel in vivo technique. Blood 106, 3691-3698.
Dudzinski DM, Igarashi J, Greif D & Michel T (2006). The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol 46, 235-276.
Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL & Jain R K (2001). Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A 98, 2604-2609.
Fulton D, Gratton JP, McCabe T J, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A & Sessa WC (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597-601.
Hatakeyama T, Pappas PJ, Hobson II RW, Boric MP, Sessa WC & Durán WN (2006). Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo, J Physiol (London) 574, 275-281.
Lal BK, Varma S, Pappas PJ, Hobson RW 2nd & Durán WN (2001). VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc Res 62, 252-262.
Kurose I, Wolf R, Grisham MB, Aw TY, Specian RD & Granger DN (1995). Microvascular responses to inhibition of nitric oxide production: role of active oxidants. Circ Res 76, 30-39.
Ramírez MM, Quardt SM, Kim D, Oshiro H, Minnicozzi M & Durán WN (1995). Platelet-activating factor modulates microvascular permeability through nitric oxide synthesis. Microvasc Res 50, 223234.
Sánchez FA, Savalia NB, Durán RG, Lal BK, Boric MP & Durán WN (2006). Functional significance of differential eNOS translocation. Am J Physiol: Heart & Circ Physiol 291, H1058-H1064.
Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW & Lisanti MP (2002). Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 277, 40091-40098.
Yuan Y, Granger HJ, Zawieja DC & Chilian WM (1993). Histamine increases venular permeability via a phospholipase C-NO synthaseguanylate cascade. Am J Physiol 264, H1734-H1739.
Yuan SY (2000). Signal transduction pathways in enhanced microvascular permeability. Microcirc 7, 395-403.