
Physiology News Magazine

Reduced mitochondrial efficiency: dysfunction or defence in ageing muscle?
Mitochondrial oxidative phosphorylation has been found to become less efficient with age in both mouse and human skeletal muscle. Here, David Marcinek explores mechanisms that can lead to either a protective or pathological effect of this uncoupling
Features
Reduced mitochondrial efficiency: dysfunction or defence in ageing muscle?
Mitochondrial oxidative phosphorylation has been found to become less efficient with age in both mouse and human skeletal muscle. Here, David Marcinek explores mechanisms that can lead to either a protective or pathological effect of this uncoupling
Features
David J Marcinek
Department of Radiology, University of Washington Medical Center, Seattle, WA, USA
https://doi.org/10.36866/pn.63.30

Mitochondria are primary sites for aerobic ATP synthesis and generation of reactive oxygen species (ROS). They also play an important role in regulating cell survival. These multiple roles of mitochondria place them at the centre of the cellular mechanisms responsible for a growing number of pathological conditions. Despite an explosion of research into mitochondrial function in ageing and diseas,e there are important issues that remain unresolved. One of these is the significance of the coupling efficiency of oxidative phosphorylation (P/O).
Reduced mitochondrial coupling in ageing tissues presents an interesting paradox. Reduced coupling has been demonstrated to reduce the generation of ROS by the mitochondria. However, uncoupling with age is also associated with impaired ATP synthesis and altered cell energetics. Therefore, certain changes associated with reduced coupling, such as reduced ATP levels, may be detrimental to cell survival, while others, like a decrease in ROS production, may be an adaptive response to cellular stress. Here I focus on mechanisms and significance of reductions in mitochondrial coupling efficiency in ageing muscle.
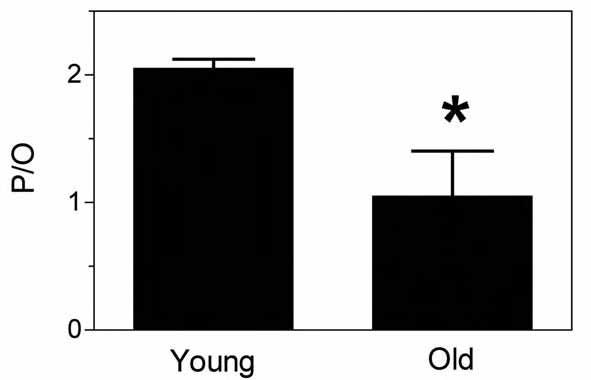
We have recently demonstrated significant mitochondrial uncoupling in vivo in aged mouse skeletal muscle (Marcinek et al. 2005). Using a combination of magnetic resonance and optical spectroscopy to directly measure ATP use and O2 uptake in vivo, we found that mitochondria in aged mouse skeletal muscle produce on average about 50% fewer ATP molecules per O2 molecules consumed (reduced P/O) than those in young muscle (Fig. 1). This mitochondrial uncoupling is consistent with the reduced in vivo phosphorylation capacity per mitochondrial volume found in the quadriceps from elderly humans (Conley et al. 2000). Mitochondrial ATP synthesis is coupled to oxygen consumption in the mitochondria through the membrane potential generated by proton pumping in the electron transport chain (ETC) (Fig. 2). This membrane potential provides the driving force for protons to flow back into the matrix, which drives ATP synthesis by complex V (F1F0 ATP synthase). Protons may also bypass the ATP synthase and leak across the inner mitochondrial membrane (IMM), short-circuiting the coupling of ATP synthesis and O2 consumption. This leak will dissipate the membrane potential and reduce the efficiency of oxidative phosphorylation (lower P/O).

Increased proton leak is the most well accepted mechanism to explain the lower P/O values in aged muscle. This hypothesis is supported by results demonstrating increased proton leak in mitochondria isolated from aged rodent muscle (Harper et al. 2004). Such proton leak may occur as a result of non-specific leak across the IMM resulting from damage to membrane components in aged muscle, or the leak could be mediated through activities of specific membrane proteins, such as the uncoupling proteins (UCP). These leak pathways are not necessarily mutually exclusive, but identifying the roles of membrane damage versus UCP function may be critical to determining whether uncoupling represents mitochondrial dysfunction (damage) or a regulated cellular response to stress (UCP) in ageing muscle.
One mechanism for increased mitochondrial proton leak with age is that oxidative damage to mitochondrial proteins and lipids leads to membrane damage making the IMM more permeable to protons. The accumulation of oxidative damage to mitochondria is well documented in aging tissues and damage to membrane lipids has been shown to increase proton leak in isolated systems. Further support for a role for oxidative damage in mitochondrial uncoupling comes from transgenic mice with reduced antioxidant activities.
Mitochondria isolated from these mice had higher levels of oxidative damage and lower respiratory control ratios (state 3/state 4 respiration) (Williams et al. 1998), which is indicative of increased proton flux across the IMM. In contrast, an increase in mitochondrial proton leak with age may be due to an upregulation of UCP activity. Increased proton leak through the UCPs would lower the membrane potential and lead to reduced ROS production by the ETC, because mitochondrial ROS production is very sensitive to changes in the resting membrane potential. This means that even a small amount of proton leak due to UCP activity could lead to large decreases in ROS production.
This idea has led to the proposal that mitochondrial uncoupling is actively regulated through UCP function as a defence against oxidative damage to mitochondria. Uncoupling by UCP3 in muscle has been found to increase in response to lipid peroxides (Echtay et al. 2003). Since the lipid peroxides form in the mitochondria in the presence of increased oxidative stress their role in upregulating UCP3 activity supports a feedback model where oxidative stress leads to mild uncoupling, which in turn reduces ROS production.
The oxidative damage and regulation of UCP hypotheses are not mutually exclusive. This raises the interesting possibility of different degrees of uncoupling in aging tissues – one that may be a regulated cellular defence against oxidative damage, and one that is the result of mitochondrial damage. In the first case mild uncoupling would be due to upregulation of UCP activity in response to oxidative stress. This would create a negative feedback loop by lowering the membrane potential and reducing ROS production. Speakman et al. (2004) found that proton leak was greater in skeletal muscle mitochondria isolated from the longest-lived mice in a population. They demonstrated that UCP3mediated uncoupling accounted for a significant fraction of the difference between the longer- and shorter-lived individuals. This study provides indirect evidence for a protective effect of mild uncoupling.
In contrast, the more severe uncoupling that has been found in aged mouse (Marcinek et al. 2005) and human (Conley et al. 2000) muscles in vivo is more likely associated with mitochondrial damage and dysfunction. The presence of mitochondrial dysfunction is indicated by reductions in the cell’s energy state (ATP/ADP) and/or ATP levels that accompany the lower P/O values in both species. The change in the cellular energetics is a sign that the ability of the mitochondria to meet the energetic demand of the cell is compromised in these tissues. Because reduced ATP levels can be signal for apoptosis, the severe uncoupling may be an early step in the pathway toward tissue degeneration.
In the model presented here UCPmediated mild uncoupling acts as a regulated, adaptive response to reduce mitochondrial oxidative stress to minimize oxidative damage. When mitochondrial oxidative stress increases with age or in disease states, eventually the level of oxidative stress exceeds the ability of the cell to counteract this stress and the result is damage to mitochondrial DNA, proteins, and lipids. This damage accumulates leading to mitochondrial dysfunction, including the severe uncoupling we have identified in vivo in aging skeletal muscle. The associated disruption of cellular energetics and reduced ATP levels may then push the cell toward apoptotic cell death and lead to tissue degeneration. Under this scenario, differences in UCP expression and the degree of mild uncoupling would indicate differences in defense against oxidative damage and contribute to the variation in mitochondrial dysfunction and tissue degeneration found in different tissues.
Acknowledgements
Thanks to Kevin Conley and Martin Kushmerick for helpful comments during the preparation of this manuscript. Development of these ideas was supported by NIH grants AG00057, AG022385 and AR36281.
References
Conley KE, Jubrias SA & Esselman PC (2000). Oxidative capacity and ageing in human muscle. J Physiol 526, 203-210.
Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otin M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ & Brand MD (2003). A signaling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. Embo J 22, 4103-4110.
Harper ME, Bevilacqua L, Hagopian K, Weindruch R & Ramsey JJ (2004). Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol Scand 182, 321-331.
Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D & Conley KE (2005). Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J Physiol 569, 467-473.
Speakman JR, Talbot DA, Selman C, Snart S, McLaren JS, Redman P, Krol E, Jackson DM, Johnson MS & Brand MD (2004). Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging Cell 3, 87-95.
Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ & Richardson A (1998). Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem 273, 2851028515.