
Physiology News Magazine

Power and control: transferring energy when there is work to be done
Features
Power and control: transferring energy when there is work to be done
Features
Naveen Sharma & William C Stanley
Departments of Physiology and Biophysics, and Nutrition, Case Western Reserve University, Cleveland, USA
https://doi.org/10.36866/pn.59.29

The heart is all about power and control. The ventricles must generate sufficient power (i.e. cardiac output x developed pressure) to maintain blood pressure and organ perfusion, and this requires the controlled transfer of chemical energy to mechanical energy. This controlled transfer of energy is most evident with the transition from resting conditions to intense exercise stress: external power generation and myocardial oxygen consumption (MVO2) increase 3 to 5 fold without ATP depletion or anaerobic metabolism. This is particularly impressive in light of the fact that cardiac ATP content is ~4 µmols g-1 and the ATP turnover at rest is ~0.25 µmols g-1 sec-1, giving a mean turnover time of only ~16 sec at rest and ~3 to 5 sec during intense exercise. Simply put, production matches need, and the system is beautifully controlled.
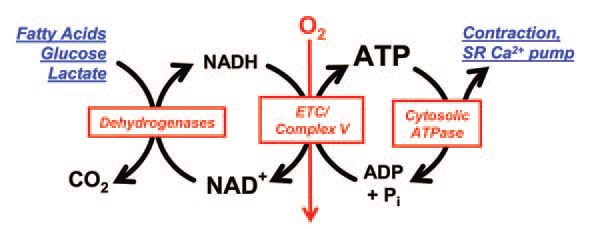
How does cardiac muscle almost instantaneously accelerate the transduction of chemical to mechanical energy? ATP breakdown in the cytosol fuels contraction and relaxation, and it is matched by the oxidation of carbon fuels in the mitochondria, the generation of NADH and aerobic ATP formation (Fig. 1). Almost all of the ATP formation in the heart comes from oxidative phosphorylation, which is driven by the activity of complex V of the electron transport chain (ETC), the proton motive force across the inner mitochondrial membrane, and the availability of ADP and inorganic phosphate (Pi). The proton gradient is fuelled by the delivery of electrons (via NADH and FADH2) to the ETC.
Studies in isolated mitochondria tell us that oxygen consumption and oxidative phosphorylation are turned on by an increase in [ADP] and the generation of NADH from carbon substrates, but what about in vivo?

It appears that [NADH] is kept constant with the transition from low to high work states due to the matching of NADH oxidation by complex I of the ETC with the rapid activation of substrate metabolism and NADH formation by pyruvate dehydrogenase (PDH), fatty acid â-oxidation, and the citric acid cycle (Fig. 2). We recently observed that with a 3-fold increase in MVO2 in pigs there was activation of PDH, maintenance of [acetyl-CoA], and an increase in the concentration of citric acid cycle intermediates (Sharma et al. 2005). In fact, even when fatty acid oxidation was almost completely suppressed by pharmacological inhibition of mitochondrial fatty acyl uptake, there were no effects on [acetyl-CoA] or [NADH], MVO2 or cardiac power. Unlike contracting skeletal muscle, the myocardium is a metabolic omnivore, able to use glucose, lactate, pyruvate, acetate, fatty acids, and ketone bodies to generate NADH. Thus there is an inherent robustness to the system, guaranteeing a sufficient supply of reducing equivalents to the ETC regardless of the metabolic milieu.
How is NADH generation activated with increased cardiac power and MVO2? While PDH can be activated by classic substrate/product regulation via decreases in NADH/NAD+ and acetyl-CoA/CoA-SH, none of these regulators are altered in the right direction when MVO2 is increased in vivo (Sharma et al. 2005). On the other hand, in vitro studies show that Ca2+ activates PDH phosphatase (which activates PDH) and the citric acid cycle enzymes isocitrate dehydronase and αketoglutarate dehydrogenase (McCormack et al.1990), stimulating NADH generation. Moreover, there is an increase in intramitochondrial [Ca2+] when there is an increase in extramitochondrial Ca2+, such as occurs with adrenergic stimulation (McCormack et al. 1990). Thus the mechanism for activation of NADH generation with increased cardiac power is feed-forward stimulation of mitochondrial dehydrogenases via the transfer of cytosolic Ca2+ to the mitochondria.
If an increase in the concentrations of [NADH] and [ADP] are not responsible for turning on flux through the ETC and oxidative phosphorylation, then what is? One possibility is that increased [Pi] concomitant with accelerated ATP hydrolysis activates the ETC and complex V. Cardiac Pi levels are normally extremely low, and early studies in isolated mitochondria showed that Pi activates mitochondrial respiration (Chance & Williams, 1956). Recent studies in isolated pig heart mitochondria show that Pi activates complex I, electron flow, and complex V (Bose et al. 2003). Another possibility is that there is direct allosteric modification of proteins in ETC complexes that effectively increase the affinity for NADH and/or FADH2. Evidence for this possibility comes from the recent observation that complex I from bovine heart mitochondria is phosphorylated in a cAMP-dependent manner (Chen et al. 2004). Further support for this concept comes from a computer model of oxidative phosphorylation, which found that only ‘parallel activation’ of ATP use and activity of the ETC complexes predicted experimental data during the rest-work transition in skeletal muscle (Korzeniewski, 2003).
There is a clear gap between our understanding of the in vivo activation of energy transduction with an increase in cardiac power, and the elegant biochemical mechanisms demonstrated in isolated mitochondria. In vivo studies are limited by the inability to measure key regulatory metabolites, particularly in the mitochondrial and cytosolic compartments, while in vitro results are generally limited by lack of physiological stimuli and inclusion of key cellular components. The goal is to understand how the system is regulated in vivo, however the system is too complex to process all of the information in one’s head. Recent publications indicate that computer modeling of myocardial metabolic systems can provide a reliable method of predicting the metabolic response to stress both in vitro (Cortassa et al. 2003) and in vivo (Zhou et al. 2005), and elucidate physiological mechanisms that are not apparent solely through experimentation. Specifically, ‘in silico’ studies allow for inclusion of fluxes within and among cellular compartments that cannot be quantified experimentally (Zhou et al. 2004). As metabolic models evolve in complexity, answers will be provided to elusive questions regarding the regulatory mechanisms that drive energy transfer in the heart under physiological and pathological conditions.
References
Bose S, French S, Evans FJ, Joubert F & Balaban RS (2003). Metabolic network control of oxidative phosphorylation: multiple roles of inorganic phosphate. J Biol Chem. 278, 39155-39165.
Chance B & Williams GR (1956). The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem 17, 65-134.
Chen R, Fearnley IM, Peak-Chew SY & Walker JE (2004). The phosphorylation of subunits of complex I from bovine heart mitochondria. J Biol Chem 279, 26036-26045.
Cortassa S, Aon MA, Marban E, Winslow RL & O’Rourke B (2003). An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J 84, 2734-2755.
Korzeniewski B (2003). Regulation of oxidative phosphorylation in different muscles and various experimental conditions. Biochem J 375, 799-804.
McCormack JG. Halestrap AP & Denton RM(1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70, 391-425.
Sharma N, Okere IC, Brunengraber DZ, McElfresh TA, King KL, Sterk JP, Huang H, Chandler MP & Stanley WC (2005). Regulation of pyruvate dehydrogenase activity and citric acid cycle intermediates during high cardiac power generation. J Physiol 562, 593-603.
Zhou L, Salem JE, Saidel GM, Stanley WC & Cabrera ME (2004). Mechanistic model of cardiac energy metabolism predicts localization of glycolysis to cytosolic sub-domain during ischemia. Am J Physiol 10.1152/ajpheart.01030.2004