
Physiology News Magazine

How mutations in myosin affect the heart
Half of all cases of familial hypertrophic cardiomyopathy are associated with a mutation in myosin. Here, Sarah Calaghan and Michelle Peckham discuss whether it is a loss or a gain of function of mutant myosin that results in hypertrophy of the heart
Features
How mutations in myosin affect the heart
Half of all cases of familial hypertrophic cardiomyopathy are associated with a mutation in myosin. Here, Sarah Calaghan and Michelle Peckham discuss whether it is a loss or a gain of function of mutant myosin that results in hypertrophy of the heart
Features
Sarah Calaghan & Michelle Peckham
School of Biomedical Sciences, University of Leeds, Leeds, UK
https://doi.org/10.36866/pn.53.24

For every 500 people reading this article, one person will have a disease known as familial hypertrophic cardiomyopathy (FHC) in which the heart is enlarged without an obvious cause, such as hypertension. It is now known that FHC is due to mutations in one of several proteins found in the cardiac muscle cell, and for a person with FHC there is a 50% chance that this mutation is in the contractile protein myosin. In this article we discuss the reasons that mutations in myosin cause enlargement of the heart (hypertrophy). Although many people with FHC have no symptoms of disease, hypertrophy places them at risk of fatal disturbances in cardiac rhythm. Indeed, FHC attracts a great deal of attention because it is the most common cause of sudden cardiac death in the young.
An introduction to myosin
Myosin is found in the thick filaments of cardiac, skeletal, and smooth muscle. It is a protein that has two globular heads about 16nm long attached to a tail that is about 160nm long (Fig. 1A). The tails pack together to form the backbone of the thick filament, and the heads stick out from the filament to interact with actin It is the interaction of myosin in the thick filaments with actin in the thin filaments that causes filament sliding, and hence muscle contraction. This interaction is driven as a result of ATP hydrolysis by myosin. After myosin hydrolyses ATP to ADP and phosphate, it binds to actin and actin accelerates the release of phosphate. Phosphate release is closely coupled to the power stroke, in which myosin heads pull on the actin filament to produce force or movement. With the arrival of numerous structures for myosin since the early 1990s, we are now starting to understand how the structure of myosin might change during the power stroke (Houdusse & Sweeney, 2001). Release of phosphate is closely coupled to the closing of the actin binding cleft as myosin forms a tight hold on actin, and to a tilting or bending of the lever which is connected to the myosin filament. The tilting of the lever is generated through part of the myosin called the converter domain (Fig. 1B), which connects the ATP pocket to the lever, and the lever rotates around a pivot found just before the converter. At the junction between the converter and the lever a few residues have a relatively unstable conformation forming a pliant region which allows the lever to tilt independently of converter movement.
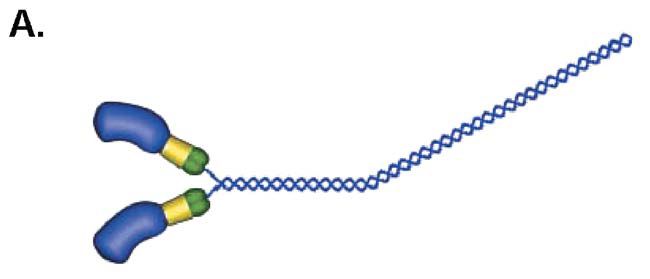
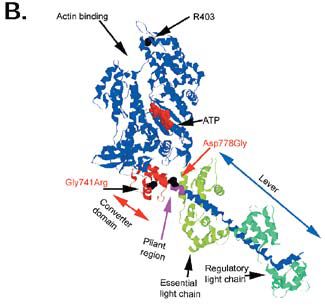
In FHC, over 50 mutant alleles have been described in the β-myosin heavy chain gene; most mutations are located in the actin- and ATPbinding sites in the motor domain, in the converter domain and in the lever arm.
Myosin in the heart
In the adult human heart, two isoforms of myosin are expressed. The predominant isoform is β-myosin heavy chain, but a small amount of α-myosin heavy chain is also present. These 2 isoforms can be distinguished on the basis of the speed with which they hydrolyse ATP; the α isoform has 2-3 times the actin-activated ATPase activity of the β-isoform. Myosin isoform expression is plastic and can change throughout life, as a result of disease, or merely as a consequence of getting older.
How do myosin mutations cause hypertrophy?
We know that mutations in myosin have negative consequences for the heart, yet, surprisingly, it is still not clear how the mutations result in cardiac hypertrophy. For example, sarcomeric disarray is known to occur in muscle that contains mutant myosin. Does this mean that mutant myosin is not incorporated normally into the sarcomere, causing aberrations of contractility that lead to hypertrophy? This possibility has been excluded because it has been shown that changes in contractility precede sarcomeric disarray in muscle expressing mutant myosin. It is now thought that it is changes in the contractility of the muscle that provide the primary trigger for the enlargement of the heart in FHC (Roberts & Sigwart, 2001).
Do myosin mutations help or hinder?
Given that changes in contractility are the primary trigger for hypertrophy in FHC, the pivotal question is whether there is a loss or gain of function of myosin. It was initially assumed that it was a depression of contractility (loss of myosin function) caused by FHC mutations that resulted in a compensatory hypertrophy (i.e. the heart gets bigger to compensate for the decline in force/power). In support of this, many of the earlier studies in this field observed a slowing of the biochemical and mechanical properties of mutant myosin. More recently, however, the notion has been raised that it may actually be hypercontractility (gain of myosin function) that causes hypertrophy as a result of the increased energy demand of the myocardium (Moss & Periera, 2000).
Take for example the Arg403Gln mutation, located in the actin binding site of the myosin head (Fig. 1B). This is the most malignant and widely studied FHC mutation in myosin. Early studies from the 1990s suggested that it is a decline in the functional properties of R403Q myosin that triggers FHC in patients with this mutation, whereas more recent work has revealed a gain in function of R403Q myosin (see Lowey, 2002 for a review). The reasons for these seemingly contradictory data may lie in the different ways that myosin function is assessed.
Ways of studying myosin function
Myosin function can be assessed in vitro, or in situ in myocytes, muscle or the intact heart. Many in vitro studies use the actin sliding filament assay, which tracks the movement of fluorescently-labelled actin filaments generated by myosin molecules immunoadsorbed onto a fixed surface. The pulling action of a single myosin molecule attached to actin can be investigated using the optical trap. Actin-activated ATPase activity can also be measured. For in vitro techniques such as these, myosin may be extracted from the muscles of patients with FHC, or from transgenic animals. More recently, mutant myosin has been produced using baculovirus/insect expression systems. There are a number of problems with these techniques. The quality and quantity of myosin preparations can have a profound effect on myosin function. In addition, many studies do not use βmyosin heavy chain (Lowey, 2002). Most transgenic work is performed with mice, which express α- rather than β-myosin. It is also much more difficult to produce striated muscle myosins using expression systems, so smooth or non-muscle myosins are used, which can differ markedly from the β-myosin heavy chain affected in FHC.
Myosin can be studied in situ by measuring parameters of contractility in myocytes, or diastolic and systolic function of the intact heart from transgenic mice. As β-myosin heavy chain is also expressed in slow twitch skeletal muscle, the contractile properties (maximum velocity of shortening, isometric force generation, power output) of skeletal muscle fibres from patients with FHC can also be used to provide an index of myosin function. One problem associated with these techniques is that there may be concomitant disorganisation of the sarcomere, or compensatory changes in other proteins besides myosin, which can complicate the interpretation of results.
A novel and direct way to look at mutant myosin function
We have recently used a novel and direct approach to examine the functional impact of mutations in myosin heavy chain. This technique has many advantages over those described above. We used myotubes, which are developing muscle fibres, formed from the fusion of mouse myoblasts (muscle precursor cells).
Myotubes do not express β-cardiac myosin, but rather a fast embryonic myosin. We grew myotubes in culture and transfected some of these with wild-type β-cardiac myosin and others with mutant β-cardiac myosin. A week after differentiation, βcardiac myosin (wild-type or mutant) is expressed at ≈25% of the total level of myosin. At this stage we looked at the effect of mutant myosin on the kinetics of contraction of myotubes stimulated at 1 Hz, and on the relationship between the frequency of stimulation and myotube shortening (see Fig. 2 for more details).
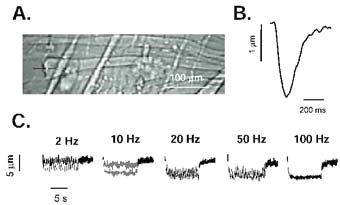
We believe that our method of assessing mutant myosin function circumvents many of the problems associated with other techniques. We are not relying on the quality of an in vitro preparation of myosin, and we are using β-cardiac myosin which is the isoform of myosin found in the human heart. In addition, from earlier work we know that the mutant myosin we are using incorporates normally into the sarcomere and does not cause sarcomeric disarray (Miller et al. 2000). It also seems unlikely that there is time for compensatory changes in proteins other than myosin to occur in these myotubes.
How do converter domain and pliant region mutations affect contractility?
To date, the hypocontractilityhypercontractility debate has focused on the FHC mutation R403Q located in the motor domain. However, there are mutations in other regions of the myosin molecule that also cause FHC. We elected to look at the impact of 2 mutations: Gly741Arg which is in the converter domain, and Asp778Gly which is at the start of the pliant region, at the junction between the converter domain and the lever arm (Fig. 1B). These mutations have been found in patients with FHC (Harada et al. 1993; Arai et al. 1995).
When we looked at the differences between myotubes expressing the wild-type β-cardiac myosin and the mutant β-cardiac myosin, it was interesting to note that, although both mutations are closely located in the myosin molecule, they have very different consequences for contraction. These are summarised in the Table. We found that the D778G mutation dramatically increased contraction kinetics and tetanic fusion frequency, compared with that seen in myotubes expressing wildtype β myosin. However, the G741R mutation did not increase the rate of contraction or relaxation at 1 Hz stimulation. There was an increase in fusion frequency with this mutation, but it was less than that seen with the D778G mutation.
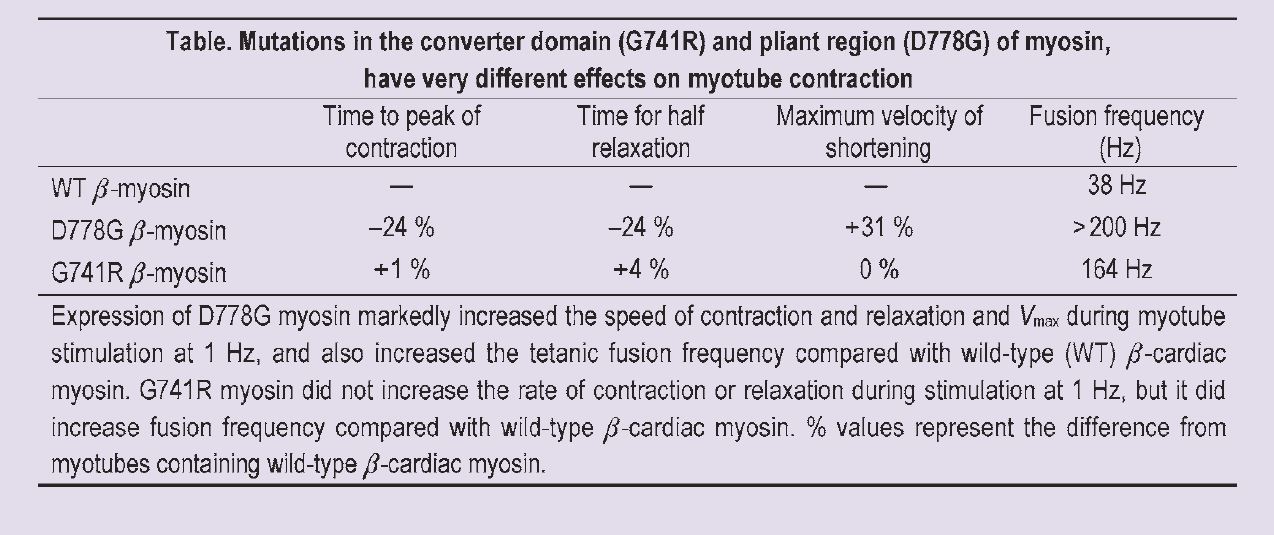
The increase in the kinetics of contraction and relaxation that we saw in myotubes expressing D778G myosin is consistent with a gain of function for myosin with this mutation. In fact the contractile parameters that we measured in myotubes expressing D778G myosin were the same as those recorded from untransfected myotubes, suggesting that the speed of D778G myosin is similar to that of the fast endogenous myosin of the myotube.
The only effect we saw in myotubes expressing G741R myosin was an increase in the tetanic fusion frequency; contractile kinetics at 1 Hz stimulation were unaffected. This shows that determination of fusion frequency is a more sensitive way to detect subtle changes in kinetics than the measurement of the rate of activation and relaxation at one frequency of stimulation. Our data suggest that the G741R mutation also causes a gain in function of myosin, but to a much smaller degree than that seen with D778G myosin.
How do our data compare with what little is known about these 2 mutations? Our results for D778G are consistent with a recent study of the equivalent mutation in expressed smooth muscle myosin which reported enhanced myosin activity (Yamashita et al. 2000). For G741R, the only report is of reduced Vmax and isometric force generation in slow skeletal muscle from FHC patients with this mutation, suggesting a loss of myosin function (Lankford et al. 1995).
To summarise
Using this novel and direct approach, we are confident of our conclusion that it is an increase in energy demand associated with hypercontractility that is the trigger for hypertrophy in FHC associated with both G741R and D778G mutations in myosin. We would predict that the phenotype of patients with mutation D778G is likely to be more severe that that of patients with mutation G741R. Interestingly, mutations in other parts of the myosin molecule, and indeed in other sarcomeric proteins, are also thought to be ‘energy cost’ diseases. Understanding the trigger for hypertrophy in FHC is one step towards finding a treatment for this disease.
Acknowledgements
The experiments detailed in this report were done in collaboration with Gaynor Miller, Joanne Maycock and Ed White from the School of Biomedical Sciences at Leeds and have been published in the Journal of Physiology (Miller et al. (2003) 548, 167-174). We’d like to thank Tim Lee for help with Figure 1, and Peter Knight for valuable discussion of myosin structure. Our work is funded by the British Heart Foundation.
References
Arai S, Matsuoka R, Hirayama K, Tamura M, Aozama T, Kimura M, Imamura S, Furutani Y, Joh-O K, Kawana M, Takao A, Hosoda S & Momma K (1995). Missense mutation of the beta-cardiac myosin heavy chain gene in hypertrophic cardiomyopathy. Am J Med Genet 58, 267-276.
Harada H, Kimura A, Nishi H, Sasazuki T & Toshima H (1993). A missense mutation of cardiac beta-myosin heavy chain gene linked to familial hypertrophic cardiomyopathy in affected Japanese families. Biochem Biophys Res Comm 194, 791-798.
Houdusse A & Sweeney HL (2001). Myosin motors: missing structures and hidden springs. Curr Opin Struct Biol 11:182-194.
Lankford EB, Epstein ND, Fananapazir L & Sweeney HL (1995). Abnormal contractile properties of muscle fibres expressing betamyosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Clin Invest 95,1409-1414.
Lowey S (2002). Functional consequences of mutations in the myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. Trends Cardiovasc Med 12, 348-354.
Miller G, Colegrave M & Peckham M (2000). N232S, G741R and D778G β-cardiac myosin mutants, implicated in familial hypertrophic cardiomyopathy, do not disrupt myofibrillar organisation in cultured myotubes. FEBS Lett 486, 325-327.
Moss RL & Periera JS (2000). Enhanced myosin function due to a point mutation causing a familial hypertrophic cardiomyopathy. Circ Res 86, 720-722.
Roberts R & Sigwart U (2001). New concepts in hypertrophic cardiomyopathies, Part 1. Circ 104, 2113-2116.
Yamashita H, Tyska MJ, Warshaw DM, Lowey S & Trybus KM (2000). Functional consequences of mutations in the smooth muscle myosin heavy chain at sits implicated in familial hypertrophic cardiomyopathy. J Biol Chem 275, 28045-28052.