
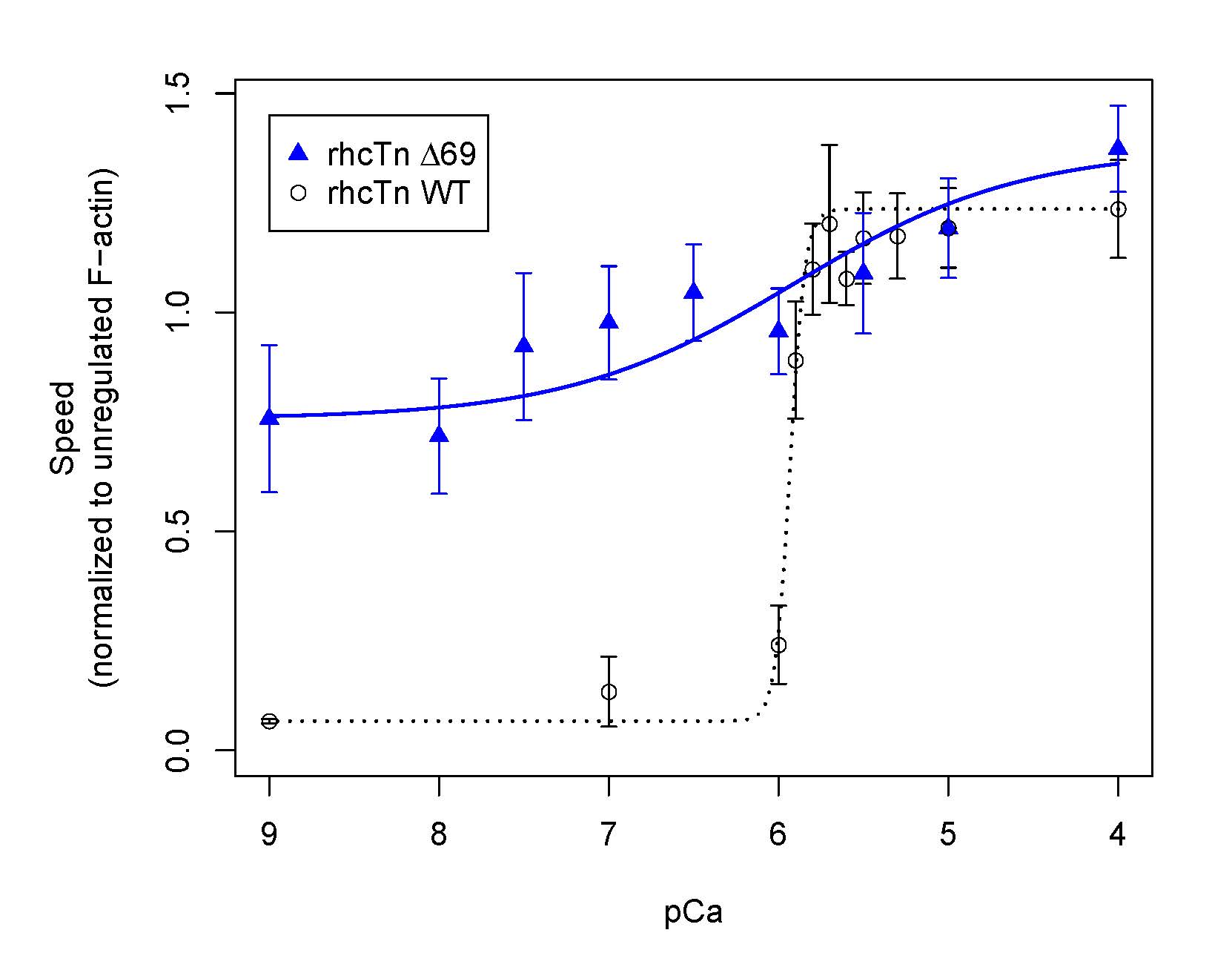
Final steps of excitation-contraction coupling in vertebrate striated muscles involve Ca2+-binding to troponin (Tn), which alters thin filament structure to allow actomyosin cross-bridge formation and sarcomere contraction in response to myocyte depolarization that transiently elevates cytoplasmic Ca2+. Published cryo-EM structures of the cardiac thin filament regulatory unit provide valuable information about Ca2+-induced structural changes in Tn and associated movement of tropomyosin (Tm) on actin, but not all portions of troponin are resolved (Yamada et al., 2020; Risi et al., 2021). We examined the functional role(s) of one region that is not fully resolved, the N-terminal “tail” of the cardiac troponin T (TnT) subunit, where T refers to Tm-binding. The tail of skeletal muscle TnT (sTnT) on its own inhibits actomyosin (Tobacman et al., 2002). Cardiac thin filament regulatory unit cryo-EM structures show, in diastolic conditions, the cTnT tail extends in one direction from the cTn core domain along actin-Tm, while the C-terminus of the troponin I (TnI) subunit (consisting of the inhibitory peptide, switch region and mobile domain) extends in the opposite direction (Yamada et al., 2020; Risi et al., 2021). Those cryo-EM structures are consistent with a model in which the cTn core binds one Tm strand while the cTnT “tail” domain reaches across the thin filament to the other Tm strand. We therefore hypothesized that the N-terminal tail of cTnT is required for full inhibition of thin filament sliding by myosin in diastolic (low Ca2+) conditions, and that apparent cooperativity of Ca2+-activation would be altered when the cTnT tail is missing. To examine the role of cTnT’s N-terminal tail, we co-expressed all three subunits of human cardiac Tn in E. coli and purified the assembled Tn complex as described (Schoffstall et al., 2011). We demonstrated through RP-HPLC purification and N-terminal peptide sequencing that the N-terminal 69 amino acids of cTnT can be removed by thrombin proteolysis at a single consensus cleavage site without affecting the other subunits. The resulting product (rhcTnΔ69) was compared in functional assays with intact cTn (rhcTn WT). Interestingly, rhcTnΔ69, unlike rhcTn WT (Schoffstall et al., 2011), does not enhance myosin (HMM) ATPase in solution in the absence of actin and Tm. Co-sedimentation demonstrates that both rhcTnΔ69 and rhcTn WT bind F-actin-Tm. In vitro motility assays of thin filament sliding were conducted as described (Schoffstall et al., 2011; Meyer & Chase, 2016). Thin filaments containing cTn missing the first 69 amino acids of cTnT only partially inhibited at diastolic Ca2+, unlike thin filaments containing rhcTn WT (left side of figure). The slope of the speed-pCa relationship was less steep (Hill coefficient < 1) for rhcTnΔ69 compared to rhcTn WT. In contrast to solution ATPase assays, maximum sliding speed at high Ca2+ levels was not reduced with rhcTnΔ69 (right side of figure). We conclude that both extensions from the core domain of cTn—the N-terminal tail of cTnT (this communication) and the C-terminus of cTnI (Meyer & Chase, 2016)—are required for full inhibition of actomyosin interactions at diastolic Ca2+ and for normal cooperative activation of actomyosin interactions.
Physiology 2021 (2021) Proc Physiol Soc 48, PC023
Poster Communications: Cardiac troponin T amino terminus is required for inhibition of Ca2+- dependent sliding of thin filaments
Linda Stroud1, Vincent A. LaBarbera1, Fang Wang1, Lauren E. Kessler1, Brenda Schoffstall2, P. Bryant Chase1
1 Florida State University, Tallahassee, The United States of America 2 Barry University, Miami Shores, Florida, The United States of America
View other abstracts by:
Where applicable, experiments conform with Society ethical requirements.