
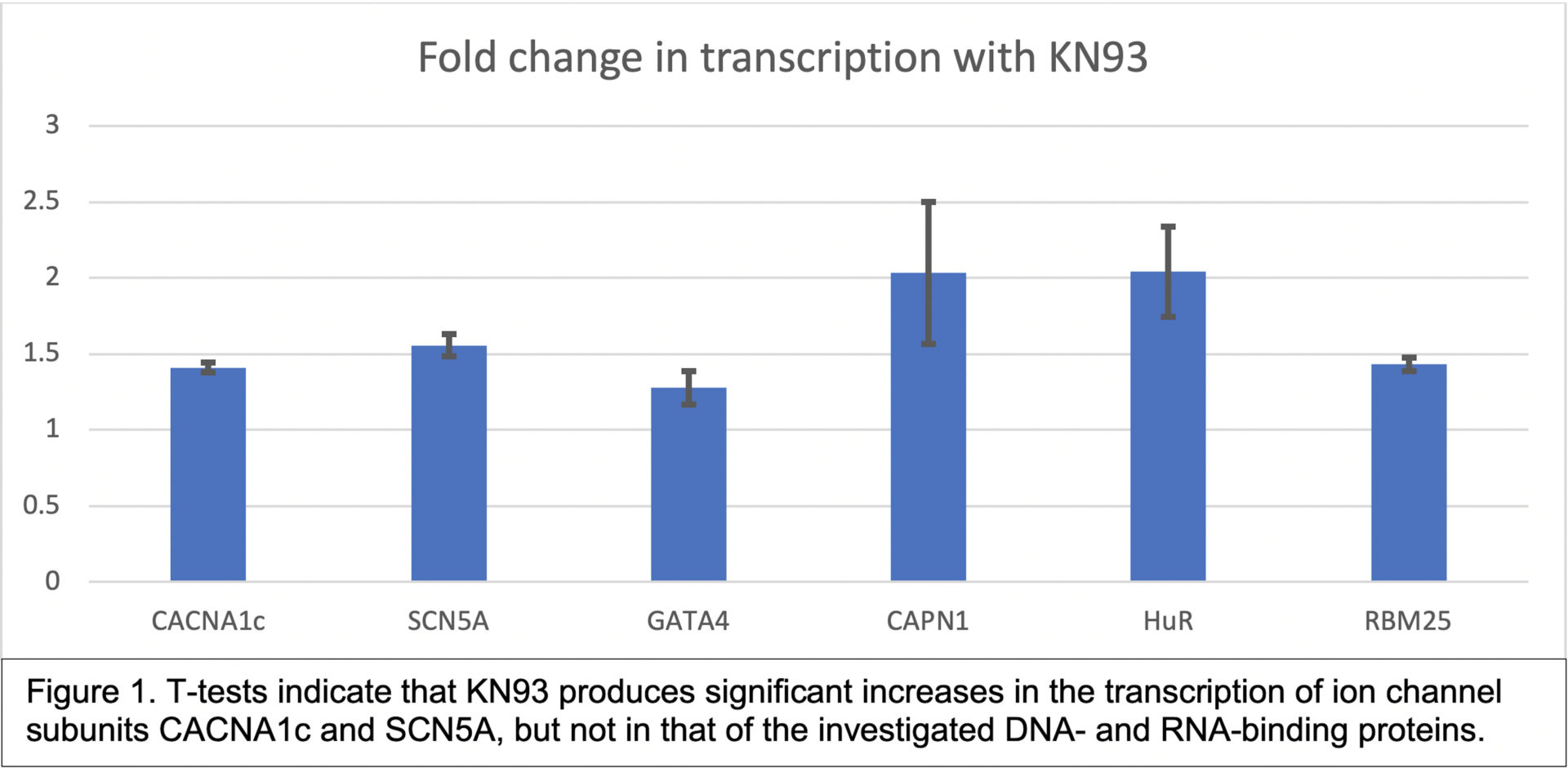
Acquired arrhythmogenesis is a pressing global health issue1, often precipitated by disturbances to the inactivation kinetics of Nav1.5, the SCN5A-encoded pore-forming α subunit of the cardiac Na+channel. CaMKII has long been known to induce such perturbations via post-translational phosphorylation2. Yet, despite the well-established secondary role of CaMKII in modifying cardiac gene expression3, less is known about whether excitation-transcription coupling extends to SCN5A. Indeed, in light of recent substantiation for both the rise in ROS-dependent CaMKII autonomy and the fall in SCN5A expression (regulated, in part, by the transcription factor GATA4) in structural heart disease, we aimed to investigate the relationship between CaMKII activity and SCN5A transcription. We thus applied the CaMK inhibitor KN934 to cultured human iPSC-derived ventricular cardiomyocytes, using its inactive analogue, KN92, and the Cav1.2-coding gene, CACNA1c, as negative and positive controls, respectively. Subsequent isolation of total cellular RNA and qRT-PCR assay enabled estimation of relative gene transcription. Unpaired, two-tailed t-tests comparing the fold changes (μ ± SEM, n= 3) in expression for CACNA1c (1.41 ± 0.03, p < 0.01) and SCN5A (1.56 ± 0.07, p < 0.05) averaged over the three cultures indicated a statistically significant increase in transcription 24 hrs following addition of either 10 or 20 mM KN93. Yet, by contrast to CACNA1c and SCN5A, the fold increase in GATA4 transcription was insignificant (1.28 ± 0.11, p > 0.05), as were the changes in expression, over the final two experiments, of DNA- and RNA-binding proteins known to influence SCN5A transcription (CAPN1: 2.03 ± 0.47, p > 0.05), pre-mRNASCN5Asplicing (RBM25: 1.43 ± 0.04, p > 0.05), and mRNASCN5Astability (HuR: 2.04 ± 0.30, p > 0.05). Over a series of three experiments, therefore, KN93 administration increased the expression of SCN5A by a similar magnitude to the positive CACNA1c control, indicating that, under normal physiological conditions, overactive CaMKII may depress SCN5A transcription in structural heart disease. However, importantly, KN93 is an inhibitor of other members of the CaMK family, including, most potently, CaMKI and CaMKIV, which have differential effects on the activities of the same transcription factors. As such, further investigations using an inhibitor that is specific to CaMKII are necessary to definitively infer the role of CaMKII from the effects of drug administration. Moreover, the lack of significant increases in transcription of the tested DNA- and RNA-binding proteins does not necessarily rule them out as mediators of the CaMKII-SCN5A relationship; Western Blot experiments are necessary to elucidate the much greater probability of their post-translational regulation5 by CaMKII-mediated phosphorylation.
Future Physiology 2019 (Liverpool, UK) (2019) Proc Physiol Soc 45, PC35
Poster Communications: Calmodulin-dependent kinase II inhibition augments SCN5A transcription in human ventricular cardiomyocytes
M. Takla1, C. Edling1, K. Jeevaratnam1
1. University of Surrey, Guildford, United Kingdom.
View other abstracts by:
Where applicable, experiments conform with Society ethical requirements.