
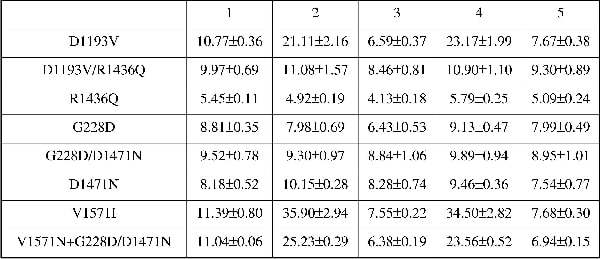
ATP-sensitive potassium channels (KATP) are present in the plasma membrane of a number of tissues and form a link between cellular metabolism and membrane excitability. This is particularly pertinent in the pancreatic β-cell where KATP channels play a major role in regulating insulin release (Ashcroft et al. 1984; Ashcroft & Ashcroft, 1990; Seino & Miki, 2003, 2004). SUR1 and Kir6.2 constitute the molecular counterparts of the channel present in pancreatic β-cells. Mutations in SUR1 and Kir6.2 occur in a hereditary disease, congenital hyperinsulinism (CHI). CHI is characterised by inappropriately high levels of insulin release and hypoglycaemia in children at birth often with disabling long-term neurological consequences. CHI is relatively rare and it is not clear how common are compound mutations and what their role is in disease pathogenesis. We have studied two families with compound mutations in the sulphonylurea receptor (SUR1) with congenital hyperinsulinism. The first patient had diffuse disease and was homozygous for two mutations in SUR1, namely D1193V and R1436Q. In a second family, the patient had focal disease and was compound homozygous for two mutations from the father (G228D and D1471N) and one from his mother (V1572I). Mutations were introduced into hamster SUR1 with site-directed mutagenesis using the QuickChange kit (Stratagene). Co-expression of mutants with Kir6.2-GFP or Kir6.2 was carried out in HEK293 cells and live cells were imaged using a Biorad Radiance 2100 laser scanning confocal Nikon TE300 microscope (Biorad, UK). 86Rb+ was used as a congener for K+ transport through the ATP-sensitive K channels in transiently transfected cells treated with or without channel stimulants and/or inhibitors. In addition, cells were studied with whole-cell patch clamp electrophysiology, under a physiological K+ gradient with 0.85 mM ATP in the pipette solution. Channel complexes containing the D1193V mutant were delivered to the plasma membrane and were functional and those containing R1436Q were also present at the plasma membrane but were non-funtional. Combining the two mutations (SUR1D1193V/R1436Q) led to intracellular retention of the channel complex. For SUR1D1193V, stimulating efflux using 100μM diazoxide or 2.5mM NaCN and 20mM 2-deoxy-D-glucose increased efflux 2-fold when compared to control (P < 0.001, one-way ANOVA with Bonferroni’s post hoc test); 10μM glibenclamide inhibited stimulation and fluxes were 0.6- and 0.7-fold lower than control, respectively (see Table 1). SUR1 G228D and D1471N singly or in combination lead to intracellular retention of the channel complex and loss of function (except for D1471N which showed activity on patch clamp). In contrast V1572 was trafficked appropriately and was functional. In addition, the expression of SUR1V1572I with SUR1 SUR1G228D/D1471N and Kir6.2 led to a recovery of function as indicated by substantial Rb+ fluxes. Our in vitro data are consistent with a mechanism in which the maternally inherited copy of SUR1 is inactivated.
University College London 2006 (2006) Proc Physiol Soc 3, PC6
Poster Communications: Compound mutations can dictate disease mechanism in congenital hyperinsulinism
Morris Chivwaba Muzyamba1, Tabasum Farzaneh1, Philippe Behe1, Khalid Hussain2, Andrew Tinker1
1. Medicine, University College London, London, United Kingdom. 2. Institute of Child Health, University College London, London, United Kingdom.
View other abstracts by:
Where applicable, experiments conform with Society ethical requirements.