
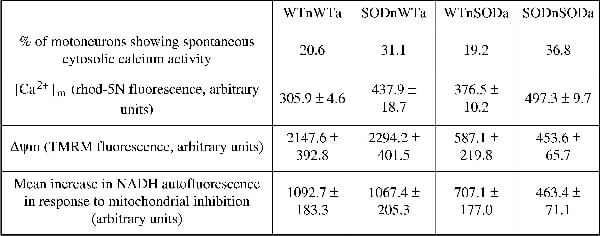
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by the selective loss of motoneurons in the spinal cord, brainstem and motor cortex. Despite extensive research, the pathogenic mechanisms underlying motoneuron degeneration are far from understood. However, increasing evidence suggests that alterations in non-neuronal cell function are likely to contribute to the process of motoneuron degeneration (Gong et al. 2000; Pramatarova et al. 2001; Clement et al. 2003). In this study, the influence of glial cell genotype on motoneuron physiology was examined at a cellular level in an in vitro co-culture model based on the SOD1 mutation. Primary motoneurons from wild-type (WT) or transgenic mice carrying the SOD1G93A mutation were plated onto a layer of either WT or mutant SOD1G93A astrocytes. Various aspects of motoneuron physiology were investigated at 7 days in vitro using confocal microscopy. Data were analysed using a one-way ANOVA incorporating a Student Newman Keuls multiple comparison test. Under resting conditions, the expression of mutant SOD1G93A in either neurons or astrocytes was associated with increased spontaneous calcium activity of co-cultured motoneurons. This increase in the frequency of transient elevations in cytosolic calcium, was consistently accompanied by a significant elevation in mitochondrial calcium concentration, [Ca2+]m, in mutant SOD1G93A expressing co-cultures (p<0.005; Table 1). The elevation in [Ca2+]m alone did not correlate with a change of mitochondrial potential (Δψm). However, mitochondria of either WT or SOD1G93A-expressing motoneurons showed a reduction in Δψm if co-cultured with mutant SOD1G93A expressing astrocytes (p<0.005). In contrast, Δψm in SOD1G93A expressing motoneurons co-cultured with WT astrocytes did not differ from WT co-cultures (p>0.3), despite an elevated [Ca2+]m (Table 1). The presence of mutant SOD1G93A-expressing astrocytes was associated with functional alterations in mitochondrial redox state in co-cultured motoneurons, as measured by NADH autofluorescence (Table 1), indicating impaired respiration, which may mediate the mitochondrial depolarisation observed in these co-cultures. These results suggest that the expression of mutant SOD1G93A in astrocytes has a significant impact on mitochondrial function of motoneurons. This alteration in mitochondrial function may increase the intrinsic vulnerability of motoneurons to the neurotoxic mechanisms proposed to be involved in ALS pathogenesis.
University College London 2006 (2006) Proc Physiol Soc 3, C86
Oral Communications: Expression of ALS-related mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria
Lynsey Bilsland1, Niran Nirmalananthan3, Linda Greensmith3, Michael R Duchen2
1. Molecular Neuropathobiology, Cancer Research UK, London, United Kingdom. 2. Physiology, University College London, London, United Kingdom. 3. Sobell Department of Motor Neuroscience and Movement Disorders, University College London, London, United Kingdom.
View other abstracts by:
Where applicable, experiments conform with Society ethical requirements.