It is generally agreed that increased cholinergic tone is an important factor in bronchoconstriction in chronic obstructive airways disease (COPD). Despite this, the mechanisms underlying acetylcholine-induced contractions are incompletely understood. Cholinergic contractions are widely thought to be mediated by postjunctional M3 muscarinic receptors (M3R) on airway smooth muscle (ASM), even though they are outnumbered 4:1 by postjunctional M2 receptors (M2R) in most species. M2R are also expressed prejunctionally on parasympathetic nerves, where they inhibit acetylcholinerelease1. Pharma companies have therefore sought to develop M3R-selective anticholinergic drugs to treat COPD because of the risk that blocking prejunctional M2R could increase acetylcholine release and exacerbate bronchoconstriction1.
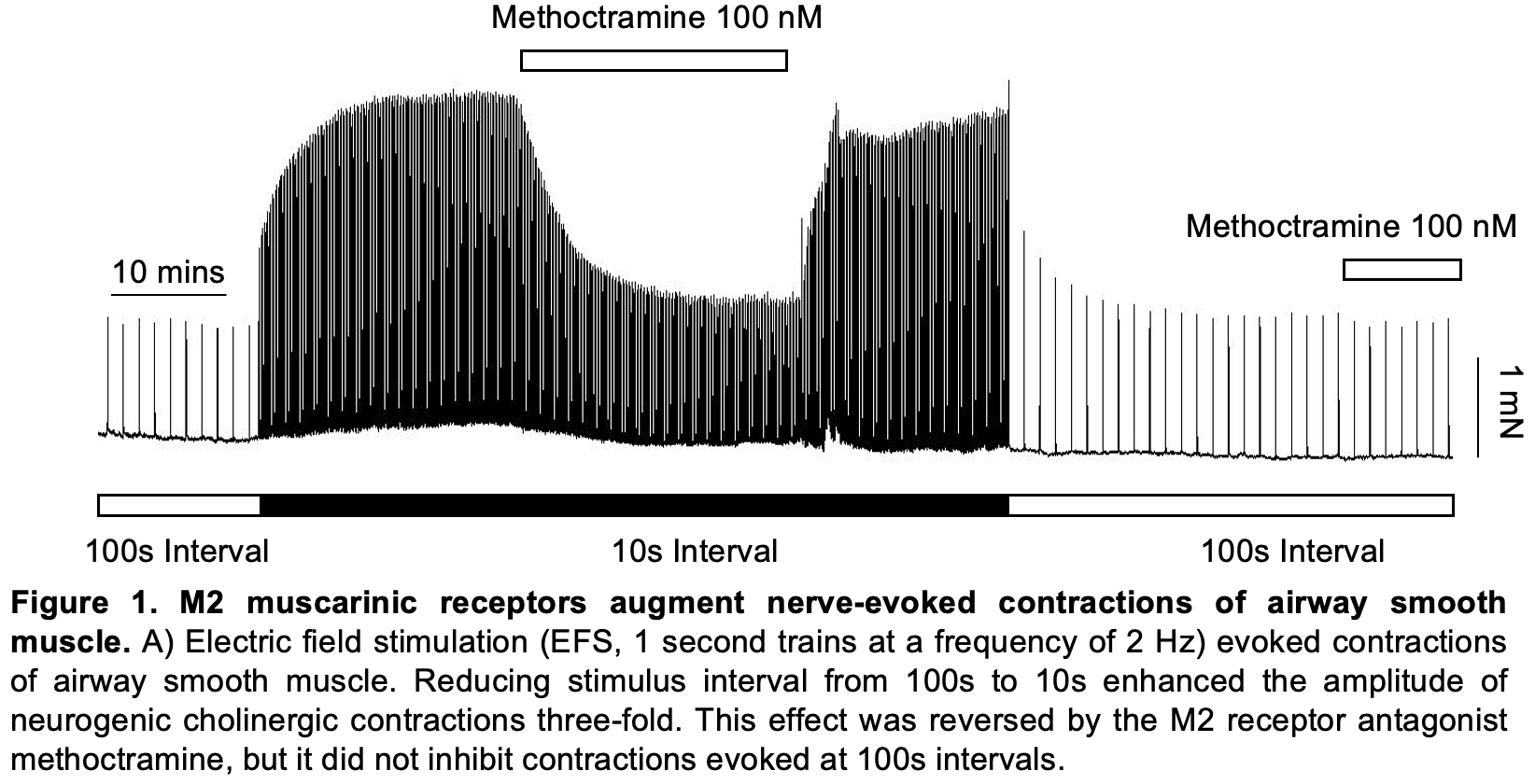
Recently, we have discovered that M2R can contribute more to cholinergic contractions than previously thought4. When mouse bronchial rings were subjected to electrical field stimulation (EFS) at 100s intervals (2Hz, 1s trains), phasic contractions were evoked that were entirely blocked by the M3R blocker, 4-DAMP and were unaffected by M2R blockers. Surprisingly, when the stimulus interval was shortened to 10s, the contraction amplitude increased by 2-3-fold. Again, the contraction was completely blocked by 4-DAMP, but this time M2R blockers (methoctramine, AFDX-116), selectively blocked all of the increase in amplitude at 10s stimulus intervals (Fig.1). Taken together, this suggested that shortening the stimulus interval resulted in M2R sensitisation of M3R-mediated contractions. When similar experiments were carried out in M2R knockout mice, shortening the stimulus interval failed to enhance contraction amplitude. Interestingly, when a sub-threshold concentration of carbachol was applied to tissues receiving the stimulus trains at 100s intervals, there was a methoctramine-sensitive increase in the amplitude of the phasic contractions in wild-type mice, but not in M2R knockout mice. This is consistent with carbachol stimulating extra-junctional M2R.
ASM cells possess L-type voltage-dependent Ca2+ channels (L-VDCC) and the means to regulate membrane potential via TMEM16A channels and various K+ channels. However, there is a lack of consensus as to the roles played by these channels in ASM contraction2,3. We further explored the mechanisms underlying the M2R-mediated enhancement of EFS-evoked contractions using blockers of either L-VDCC (nifedipine, verapamil) or TMEM16A (Ani9, CaCCinhA01). These drugs all failed to affect responses evoked at 100 s intervals, but completely reversed the enhancement seen on switching to 10s stimulation. Thapsigargin, a SERCA pump blocker, enhanced EFS responses at 100s intervals in a similar manner to switching to 10s intervals, and this effect was reversed by nifedipine or Ani9.
We propose a model whereby cholinergic stimulation results in L-VDCC activation by TMEM16A-mediated membrane depolarisation. In M3R-exclusive responses, the incoming Ca2+ is immediately taken up into sub-membrane sarcoplasmic reticulum via SERCA (the ‘superficial buffer barrier’ mechanism). When M2Rs (possibly located extra-junctionally) are co-activated, Gi protein-mediated suppression of cAMP production results in SERCA pump inactivation, which allows the incoming Ca2+ to reach the contractile proteins. Since extra-neuronal production of acetylcholine has been proposed to contribute to increased cholinergic tone in COPD5, we suggest that this may stimulate M2R on ASM and contribute to the bronchoconstriction typical of this disorder.
All work was carried out in compliance with EU legislation.