
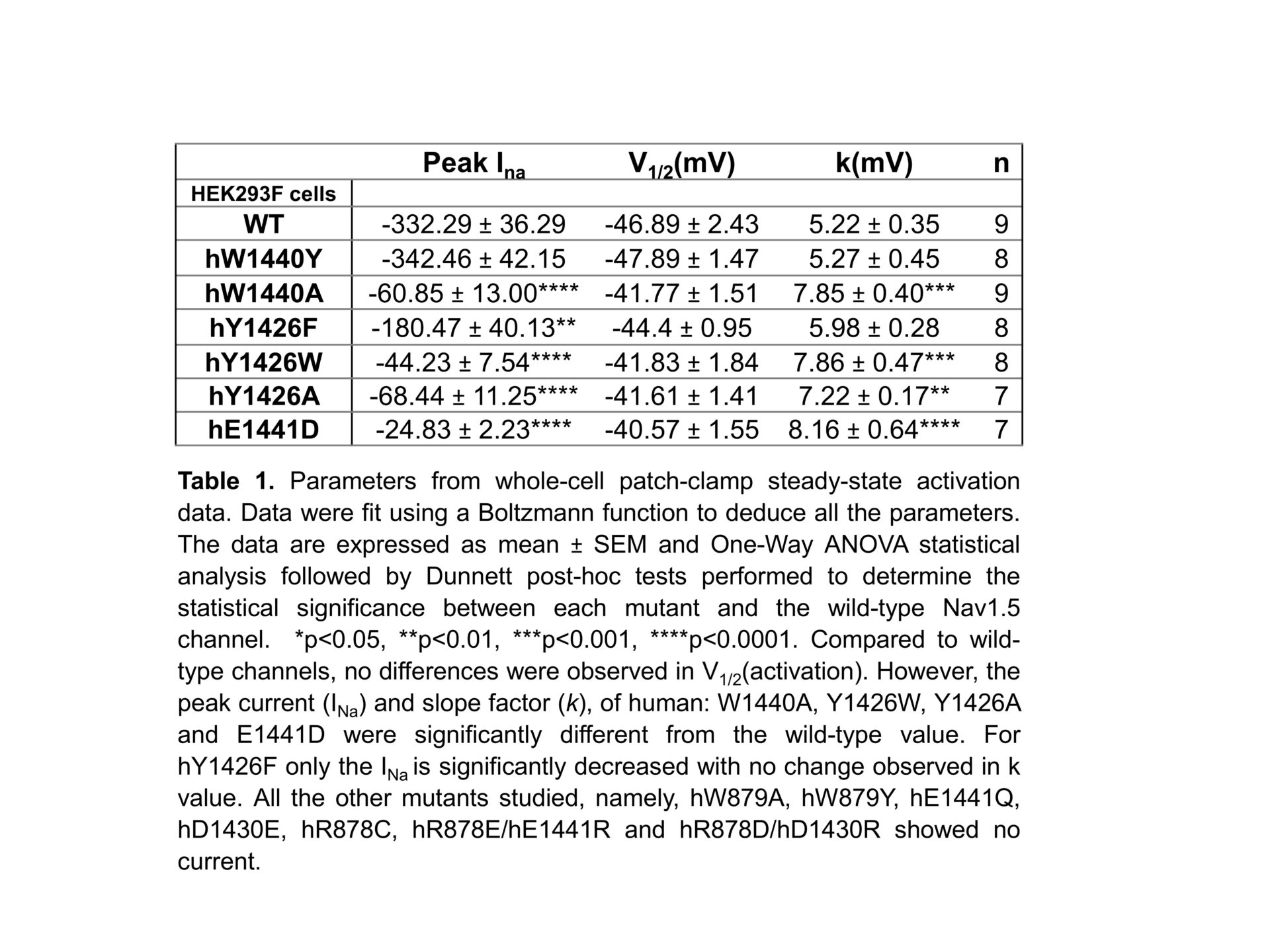
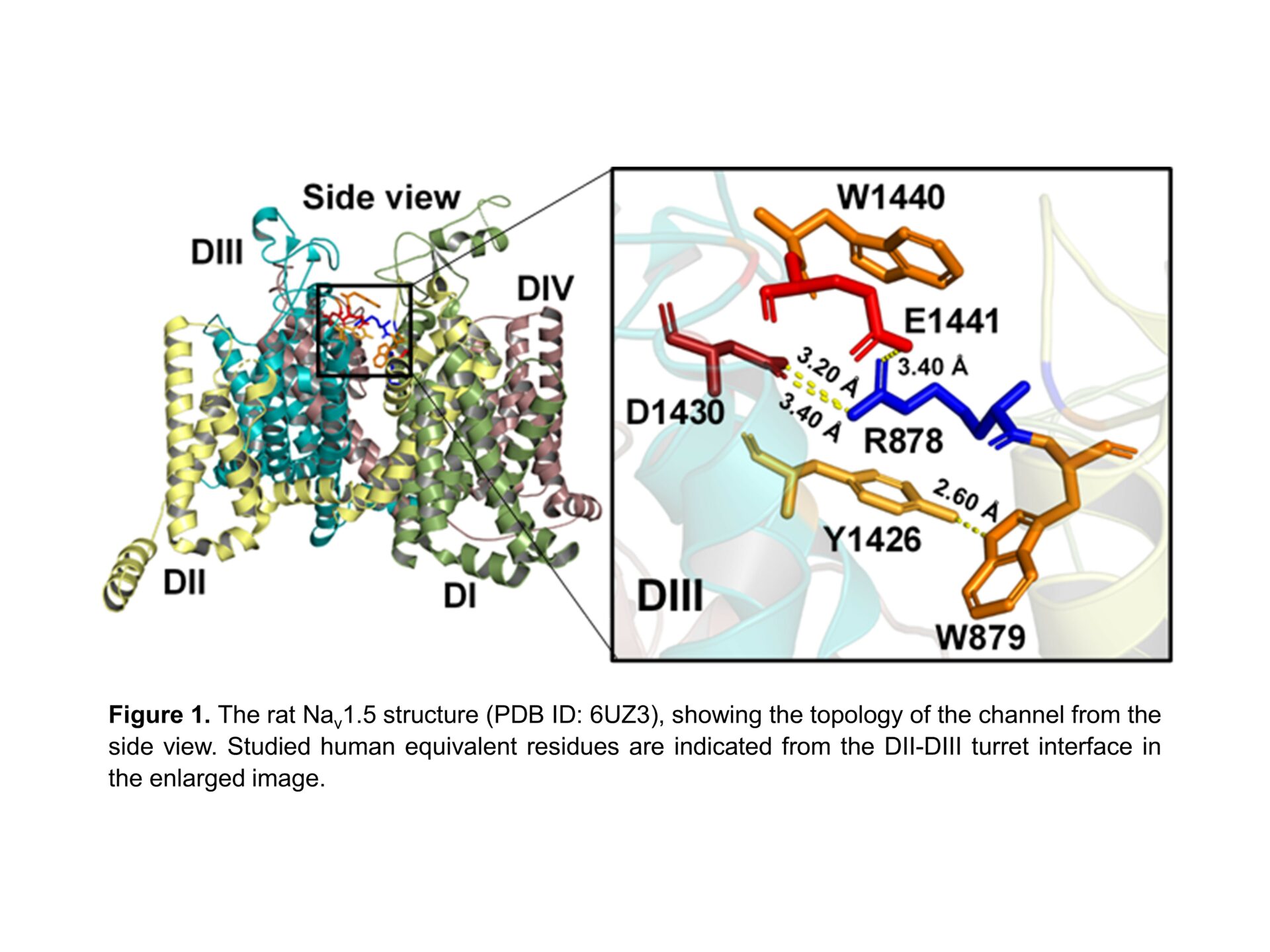
The cardiac voltage-gated sodium channel, Nav1.5, is central to cardiac action potential initiation and therefore cell membrane depolarization. The extracellular turrets of the channel extend over the central pore and play a role in ion selectivity and permeation1. Several germ-line mutations within this region have been implicated in cardiopathologies such as Brugada Syndrome (BrS), yet it is unclear how they affect channel gating2,3. From the recent cryogenic electronic microscopy structure of mammalian Nav1.5 (PDB ID:6UZ3) 4, these mutants appear to form a complex salt bridge between an arginine residue (R878) and two acidic residues (D1430 and E1441) that connect the channel turret regions of the DII and DIII domains (Figure 1). Furthermore, the complex salt bridge is bolstered with adjacent cation-π interactions with the aromatic residues (W879, Y1426 and W1440). We employed site-directed mutagenesis to systematically examine this region. We transiently transfected mutants into HEK293F cells to study their gating behaviour (steady-state activation, inactivation, and recovery from inactivation protocols) by whole-cell patch clamp and performed One-Way ANOVA followed by Dunnett post-hoc tests with results displayed as mean±SEM. We also performed in silico Molecular Dynamic (MD) simulation modelling to investigate the structural stability of the mutants relative to the wild-type channel. Almost all the generated mutants displayed either reduced or no detectable peak currents. Nevertheless, surface biotinylation demonstrated that all the mutants were expressed on the plasma membrane, therefore ruling out defective trafficking. Nearly 80% of our mutants that displayed no detectable gating activity were targeted to the complex salt bridge. These included all the natural BrS mutants that have been previously identified in this region. Of the mutants that retained some gating activity, 83% were targeted to the cation-π bonds. Thus, the complex salt-bridge may provide the major stabilisation to the DII-DIII turret interface, whilst the cation-π bonds contributes secondary – although still important functional reinforcement. The Nav1.5 activator, veratridine, failed to rescue any of the null-mutants. For the mutants that retained some gating activity, only peak currents and steady-state activation parameters were perturbed relative to the wild-type channel (Table 1). Steady state inactivation parameters and recovery from inactivation kinetics were not affected. Taken together, these results suggest that the compromised gating behaviour of the mutants is not caused by large-scale protein misfolding. Rather, the mutations are more likely to introduce relatively subtle structural alterations, that selectively block the channel activation step. MD simulations revealed that the mutations destabilised the channel outer and inner vestibules through which the sodium ions move during channel activation. The mutations were also predicted to perturb the geometry of the DEKA selectivity ring that normally traps and concentrates sodium ions in the boundary between the outer and inner vestibules. We note that this structural feature is strongly conserved in other Nav channel isoforms and pathological mutations in the same region of Nav1.1 and Nav1.7 have previously been described in clinical literature. Thus, our findings provide general insights into a class of pathological mutations occurring not only in Nav1.5 but also in other sodium channel isoforms.
Physiology 2021 (2021) Proc Physiol Soc 48, PC004
Poster Communications: Pathological turret mutations in the cardiac sodium channel Nav1.5 induce long-range disruption to the pore geometry
Zaki F Habib1, 2, Manas Kohli2, 3, Samantha C Salvage2, Taufiq Rahman3, Christopher L-H Huang1, 2, Antony P Jackson2
1 Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, United Kingdom 2 Department of Biochemistry, University of Cambridge, Tennis Court Road, CB2 1QW, Cambridge, United Kingdom 3 Department of Pharmacology, University of Cambridge, Cambridge, United Kingdom
View other abstracts by:
Where applicable, experiments conform with Society ethical requirements.