At many central synapses, exocytotic release of GABA is modulated by the activation of presynaptic GABAB receptors. In contrast to detailed structural knowledge, GABAB receptor signalling mechanisms remain somewhat unresolved (Misgeld et al. 1995); furthermore, a more novel mechanism whereby G protein-coupled receptors (GPCRs) may directly couple to the exocytotic machinery has been shown in lamprey motoneurons (Blackmer et al. 2001). Here, I examined the major signalling pathways proposed to couple to GABAB receptor activation in mammalian central neurons by investigating the effects of GABAB receptor activation on inhibitory GABAergic transmission onto Purkinje cells.
Inhibitory postsynaptic currents (IPSCs) and miniature IPSCs (mIPSCs) were recorded using whole-cell patch-clamp recordings in parasagittal cerebellar slices, obtained from humanely killed 3- to 5-week-old mice. Data are presented as means ± S.E.M. (n), where n is the number of cells.
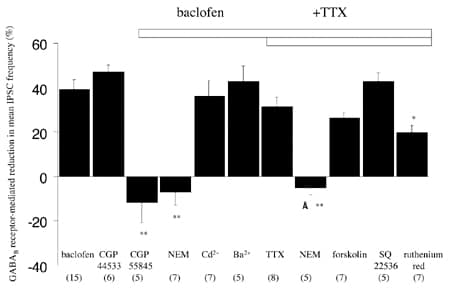
The GABAB receptor agonists baclofen and CGP 44533 (both 25 µM) caused a clear, reversible reduction in mean IPSC frequency. In subsequent experiments, the effects of a number of agents on this baclofen-induced inhibition were determined (Fig. 1). In all of these experiments, no clear effects were seen on mean IPSC (or mIPSC) amplitude, consistent with a presynaptic site of action. Baclofen inhibition was blocked by the GABAB antagonist CGP 55845 (5 µM). Baclofen effects were also occluded by N-ethylmaleimide (NEM; 50 µM), an alkylating agent which uncouples Gαo/Gαi subunits. Baclofen inhibition was unaffected by the general voltage-dependent Ca2+ channel blocker Cd2+ (50 µM) and the general K+ blocker Ba2+ (100 µM). Also, baclofen still produced a robust inhibition of mIPSCs isolated by tetrodotoxin (TTX, 1 µM). Under these conditions, the adenylate cyclase inhibitor SQ 22536 (100 µM) also did not prevent baclofen inhibition. Finally, the secretagogue ruthenium red (10 µM) was used to directly stimulate the synaptic release machinery (see Bardo et al. 2002). Importantly, baclofen was able to inhibit GABA release under these conditions, in which other downstream mechanisms were bypassed. However, baclofen inhibition was attenuated in comparison to controls (P < 0.01), which may be consistent with a convergent site of action for ruthenium red and baclofen.
This study provides evidence that GABAB receptor-mediated inhibition of GABA release onto Purkinje cells involves Gαo/Gαi (or their liberated Gβλ) subunits and that action occurs downstream of any effects on major ion conductances or adenylate cyclase. The action of baclofen in the presence of ruthenium red is consistent with a signalling pathway involving a direct interaction with the synaptic machinery in this pathway.
This work was supported by The Wellcome Trust.