
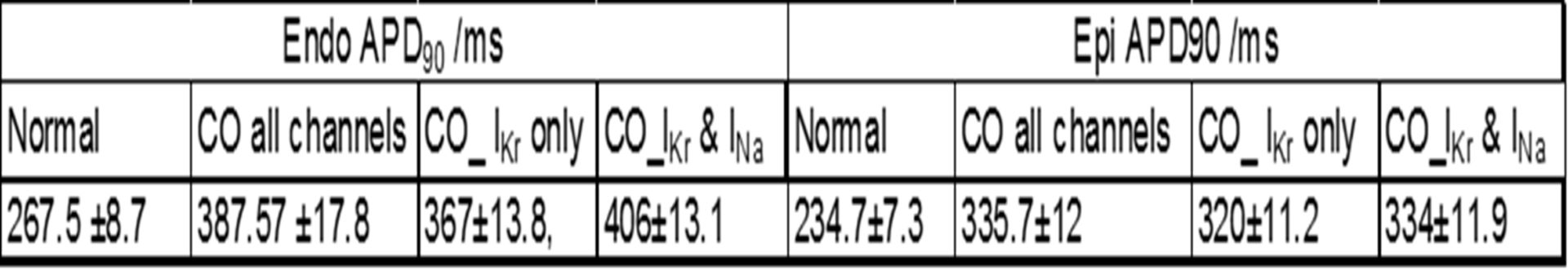
Carbon monoxide (CO) is a well-known toxin and exposure to CO has been related to cardiovascular complaints, including arrhythmias1. Previous studies have shown that CO blocks Ca2+ and Na+ currents and can induce early afterdepolarisations in rat ventricular myocytes2. We examined possible mechanisms for CO arrhythmias and the role of hERG in arrhythmogenesis in guinea pig ventricular myocytes and models of human ventricular cells and tissues. Whole-cell patch clamp recordings were made from isolated guinea pig myocytes, as well as from HEK293 cells expressing hERG gene. CO was applied via the CO-releasing molecule, CORM-2 (1-10µM). The formation of peroxynitrite (ONOO-) in HEK293 cells was monitored fluorimetrically. Values are means ±S.E.M. The effects of CO on human ventricular myocyes were modelled by -50, +30,-65 and +105 % changes in the maximal conductances for ICa, IK1, IKr and INa in the O’Hara-Rudy3 family of human ventricular cell models. Cell models and tissue models were solved with a space step of Δx=0.2mm, an adaptive time step of 0.01ms-0.25ms. Cell model conductance parameters were Gaussian distributed with a ±5% standard deviation. CO irreversibly inhibited hERG currents in HEK293 cells by 65.2 ±4.01%; (3µM; n=6, P<0.001, Student’s paired t test), the inactive form of CORM-2 had no significant effect. Current amplitudes were restored by the hERG activator, NS1643 (3µM, 90.8 ±5.5%, P< 0.001, n=7). Currents were also strongly inhibited by E4031 (75 ± 5.5 % inhibition, 3µM, n=4, P<0.001). In guinea pig myocytes action potentials were prolonged and early afterdepolarizations were observed. Inhibition was prevented by ebselen (100nM), antimycin A, or myxothiazol, 1µM), suggesting CO-mediated inhibition involves mitochondria-derived ROS. Pre-treating cells with L-NAME (1mM) abolished the inhibitory effects of CORM-2, indicating CO inhibition requires Nitric oxide (NO) formation. CO raised ONOO- levels, an effect reversed by ONOO- scavenger FeTPPS, preventing inhibition of hERG tail currents, and abolishing the CO effects on APs in guinea pig myocytes4. Computational modelling by incorporating the CO-induced conductance changes into human cell models showed that with 5% variability, n=500, in all the maximal conductance parameters, the endo- and epi-cardial APD90 and its variability is increased at a BCL of 1000ms, see table 1 for details. Prolongation of the APD90 is seen at all BCL, CO abolishes alternans in the endo-, and induces alternans in the mid-myocardial cell models. The dispersion of repolarisation and the propagation velocity in ventricular tissue and wall models is decreased, and the vulnerable window is increased5.These data suggest that CO effects guinea pig cardiac myocytes via the ONOO- mediated inhibition of hERG channels. This inhibition is pro-arrhythmogenic in tissue models, by action potential prolongation, increase in dispersion of refractoriness, and an increased vulnerability to re-entry.
Physiology 2019 (Aberdeen, UK) (2019) Proc Physiol Soc 43, PC042
Poster Communications: The Role of Human Ether-a-go-go-Related Gene K+ Channels in Cardiac Arrhythmias induced by Carbon Monoxide
M. Al-Owais1, N. Hettiarachchi1, D. Steele1, C. Peers1, A. Holden1, A. Benson1
1. University of Leeds, Leeds, United Kingdom.
View other abstracts by:
Where applicable, experiments conform with Society ethical requirements.