Introduction
CFTR is an anion-selective channel which plays important roles, regulating fluidity and pH of luminal secretions. New modulator drugs targeting CFTR have highlighted previously unexpected involvement of CFTR in physiological and pathological processes [1-4]. CFTR-targeting drugs also have the potential to help improve our mechanistic understanding of CFTR gating function and biogenesis, especially when structural details of binding are known [5, 6].
To improve our molecular understanding of CFTR function and pharmacology we tested compounds known to inhibit MsbA and P-glycoprotein, both Type IV ABC transporters [7], like CFTR. Quinoline inhibitors bind on MsbA at positions homologous to CFTR’s portals. In the open channel conformation CFTR’s portals are thought to be part of the permeation pathway connecting the inner pore vestibule to the cytosol. Inhibitors Elacridar and Zosuquidar occupy the substrate-binding cavity in P-glycoprotein, structurally homologous to CFTR’s inner vestibule.
Methods
We tested the ABC transporter inhibitors on CFTR using a high-content fluorescence assay [8]. YFP-CFTR, an N-terminal fusion of an iodide-sensitive YFP [9] to CFTR, was coexpressed in HEK293 cells with a cytosolic mCherry. Exploiting a microscopic image-acquisition system and watershed image-segmentation algorithms (relying on mCherry fluorescence), we monitored the time course of YFP quenching in individual cells, following extracellular iodide addition. The amount of iodide entering each cell was estimated from the integral of the relative YFP fluorescence decrease following iodide addition (area above the curve over 39 s, AAC39), providing a robust readout of CFTR activity.
Results
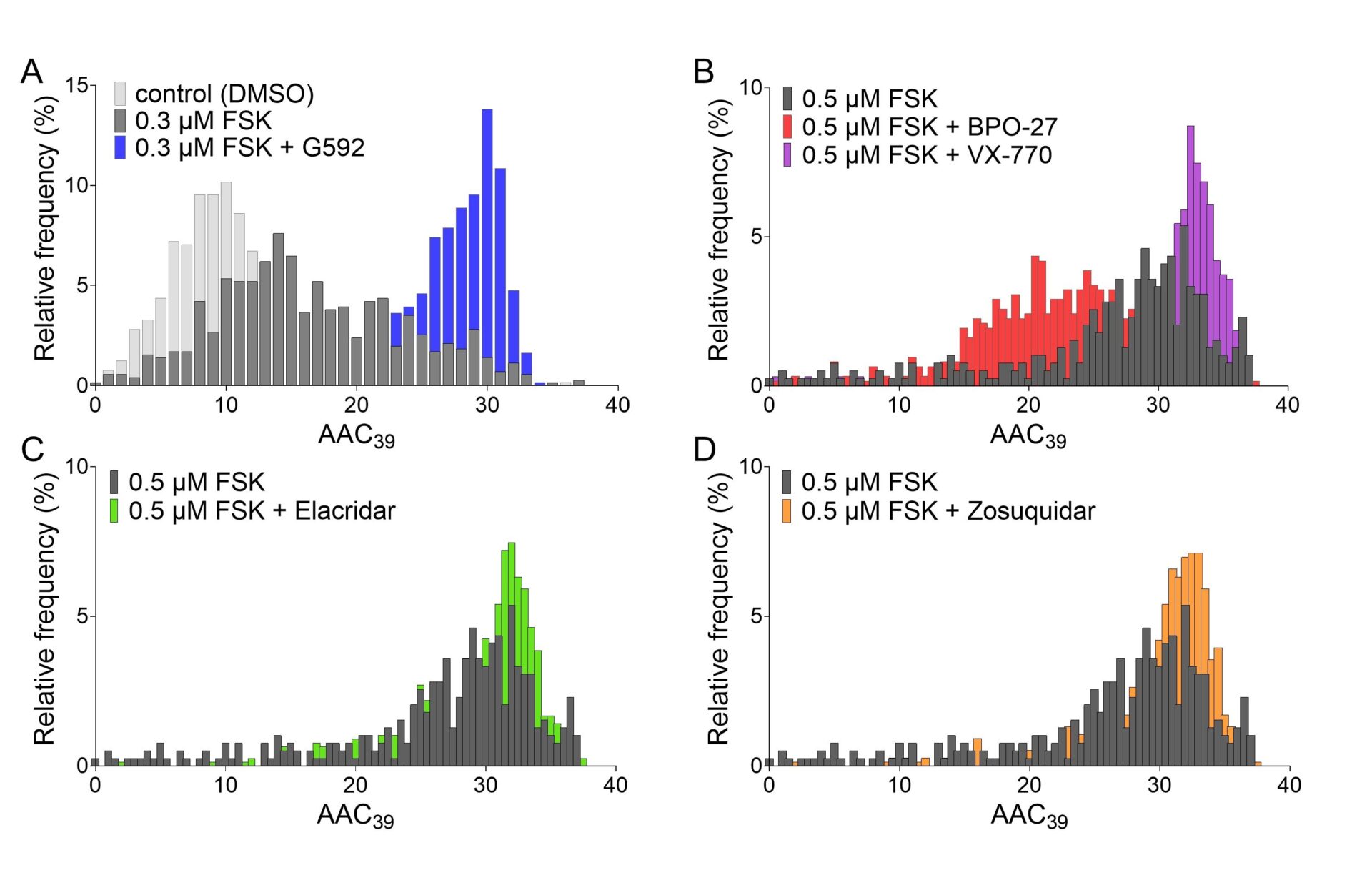
Upon activation of CFTR by addition of forskolin (at EC50, 0.3 μM), frequency distributions of the AAC39 metric shift towards higher values (figure 1A), reflecting increased PKA-dependent CFTR phosphorylation. In the presence of 0.5 μM forskolin, 10 μM BPO-27 or 3 μM VX-770, CFTR-specific inhibitor and potentiator, respectively, shifted AAC39 distributions to lower and higher values, as expected (figure 1B). Activation of CFTR in the presence of inhibitors of MsbA (10 μM G592 (S)-enantiomer [10], figure 1A) or P-glycoprotein (10 μM Elacridar, figure 1C, or 10 μM Zosuquidar, figure 1D) resulted in more cells populating histogram bins corresponding to larger integral areas above the quenching curve. Activation in the presence of G593 ((R)-enantiomer) resulted in a significantly smaller AAC39 (0.3 μM forskolin + G592: 25.02±0.90 RFU*s (n=6) vs. 0.3 μM forskolin + G593: 22.24±0.89 RFU*s (6), P=0.039, paired t-test) inconsistent with non-specific action at the membrane [10]. Moreover, the activating effect of Elacridar or Zosuqidar disappeared when P-glycoprotein inhibitors were combined with VX-770 (0.5 μM forskolin + VX-770: 29.68±1.33 RFU*s (4); 0.5 μM forskolin + VX-770 + Elacridar: 28.03±1.03 RFU*s (4); 0.5 μM forskolin + VX-770 + Zosuquidar: 28.58±1.35 RFU*s (4)). The lack of additivity suggests that both potentiator and P-glycoprotein inhibitors bind to CFTR.
Conclusion
P-glycoprotein and MsbA are ATPase-driven pumps, while hydrolysis of bound ATP controls the rate of channel closing in CFTR. Obstructing the ATP hydrolysis step could inhibit substrate transport in pumps and increase open probability in CFTR. Yet why the binding of compounds at CFTR’s portals and inner vestibule increases cellular anion conductance, rather than hinder anion permeation, remains unclear.