
Physiology News Magazine

A starring role for astrocytes
Recent evidence continues to re-cast the role of astrocytes in brain function. Astrocytes now appear to express a rich repertoire of signalling mechanisms, can respond to neuronal signals and rapidly signal back to neurons. The recent work of Stephen Traynelis and his colleagues has further emphasized the ability of astrocytic receptors to control excitatory synaptic transmission. Thus these cells have emerged as active participants in brain function
Features
A starring role for astrocytes
Recent evidence continues to re-cast the role of astrocytes in brain function. Astrocytes now appear to express a rich repertoire of signalling mechanisms, can respond to neuronal signals and rapidly signal back to neurons. The recent work of Stephen Traynelis and his colleagues has further emphasized the ability of astrocytic receptors to control excitatory synaptic transmission. Thus these cells have emerged as active participants in brain function
Features
Guido Mannaioni (1), Hongjie Yuan (2), C Justin Lee (3), & Stephen F Traynelis (2)
1: Department of Pharmacology, University of Florence, Florence, Italy
2: Department of Pharmacology, Emory University School of Medicine, Atlanta, GA, USA
3: Center for Neural Science, Division of Life Sciences, Korea Institute of Science and Technology, Seoul Korea
https://doi.org/10.36866/pn.70.31

As recently as two decades ago, the primary function of astrocytes was still sometimes mistakenly described as only a supportive matrix for the brain’s neurones. Although the word astrocyte means ‘star’ shaped cell, astrocytes were certainly not thought to play a starring role in central nervous system function at that time. Rather, they were typically assigned a number of supportive and nutritive roles. Over the years, this perception has gradually shifted. Today (more in line with their name), astrocytes appear to take part as primary actors in the regulation of synaptic transmission, a process known to be crucial to the biological computations that underlie thought and perception. Astrocytes can respond to neurotransmitters used in synaptic transmission, clear neurotransmitters from the extracellular space to terminate synaptic signaling, and fine-tune the strength of connections through a variety of different mechanisms. Thus, there is now clear appreciation of the more active and important roles being considered for astrocytes.
The first clue about the communication between astrocytes and neurones was noted in the 1980s when it was found that astrocytes could respond to the chemical signals sent by neurones. Astrocytes express on their cell membrane a large number of neurotransmitter receptors, and these can be activated by neurotramsitters released from neurones. A major step forward occurred in 1994 when Parpura and co-workers showed for the first time that astrocytes were able to signal back to neurones (Parpura et al. 1994). Indeed, when single astrocytes were directly stimulated to increase internal calcium and release glutamate, calcium levels of adjacent neurones were increased. Since this discovery, multiple lines of experimentation have defined numerous examples of neurone-astrocyte and astrocyte-neurone communication. However, the molecular mechanisms by which neurone-astrocyte communication takes place as well as its specific role in physiology and/or pathology remain an active topic of investigation. One particularly interesting set of results suggest that G-protein-coupled receptors on the cell surface of astrocytes can stimulate release of Ca2+ from intracellular storage sites. Several lines of evidence suggest that the subsequent elevation of intracellular Ca2+ levels can trigger release of the neurotransmitter glutamate from astrocytes by a number of different processes (e.g. Parpura et al. 1994; Bezzi et al. 1998; Araque et al. 2000). Glutamate released in this manner is estimated to reach concentrations between 1-6 micromolar, enough to activate neuronal glutamate receptors and influence neuronal function (Lee et al. 2007).
Even though the Ca2+-dependent release of glutamate from astrocytes is now a well-documented phenomenon, both the mechanism and physiological roles are still debated. Two classes of mechanisms that have been proposed for astrocytic glutamate release include regulated vesicle exocytosis and channel-mediated glutamate release. In the first mechanism, glutamate is hypothesized to be packaged into vesicles that undergo exocytosis following an increase in intracellular Ca2+. Channel-mediated glutamate release is thought to involve glutamate permeation through channels that open in response to various stimuli.
A number of studies have suggested that glutamate released from astrocytes is capable of activating ionotropic or metabotropic glutamate receptors on neurones. Furthermore, in a recent report, activation of neuronal G-protein coupled metabotropic glutamate receptors by astrocytic glutamate release has been proposed to potentiate NMDA receptor function (Wirkner et al. 2007). This is supported by recent findings from our lab (Lee et al. 2007). Further-more, the low level activation of NMDA receptors that we report appears to potentiate synaptic NMDA receptor function by a Mg2+-dependent mechanism rather than intra-neuronal post-translational modification. By using a sensitive bio-detector of glutamate release, we demonstrate that activation of P2Y purinergic receptors, bradykinin receptors, and protease activated receptors (PARs) all stimulate glutamate release from cultured or acutely dissociated astrocytes (Fig. 1A, B). We chose protease activated receptor PAR1 as a model system to study in depth because of favorable pharmacological and molecular tools, its prominent expression in astrocytes, and its high relevance to neuropathological processes. Astrocytic PAR1-mediated glutamate release in vitro is Ca2+- dependent and activates NMDA receptors on adjacent neurones in astrocyte-neurone co-cultures. Activation of astrocytic PAR1 in hippocampal slices evokes an APV-sensitive inward current and causes APV- sensitive neuronal depolarization in CA1 neurones. PAR1 activation enhances the NMDA receptor-mediated component of synaptic miniature EPSCs recorded under voltage clamp, evoked EPSCs under voltage clamp, and evoked EPSPs under current clamp. Magnesium is a divalent ion that is capable of blocking neuronal NMDA receptors at resting membrane potential, and all forms of potentiation are Mg2+-dependent. The data described in this paper suggest that any potential neuronal depolarization mediated by dendritic NMDA receptors in response to astrocytic glutamate release, regardless of the mechanism of release, may be capable of reducing Mg2+-dependent block and potentiating the NMDA receptor component of EPSPs. A large number of G-protein-coupled receptors are coupled to release of intracellular Ca2+ from internal stores through the Gαq/11 signalling pathway. These receptors are expressed by mammalian astrocytes, and these results suggest that many of these additional receptors might be able to engage the proposed mechanisms. However, a recent study showed transgenic astrocyte-targeted expression of a peripheral G-protein-coupled receptor that mediates Ca2+ signaling did not detectably alter neuronal function. These data suggest that Ca2+ signalling in astrocytes might not be involved in synaptic modulation, and also raise a caveat regarding the transfer of conclusions reached from experiments involving activation of one astrocytic G-protein coupled receptor to other G-protein coupled receptors (Fiacco et al. 2007).
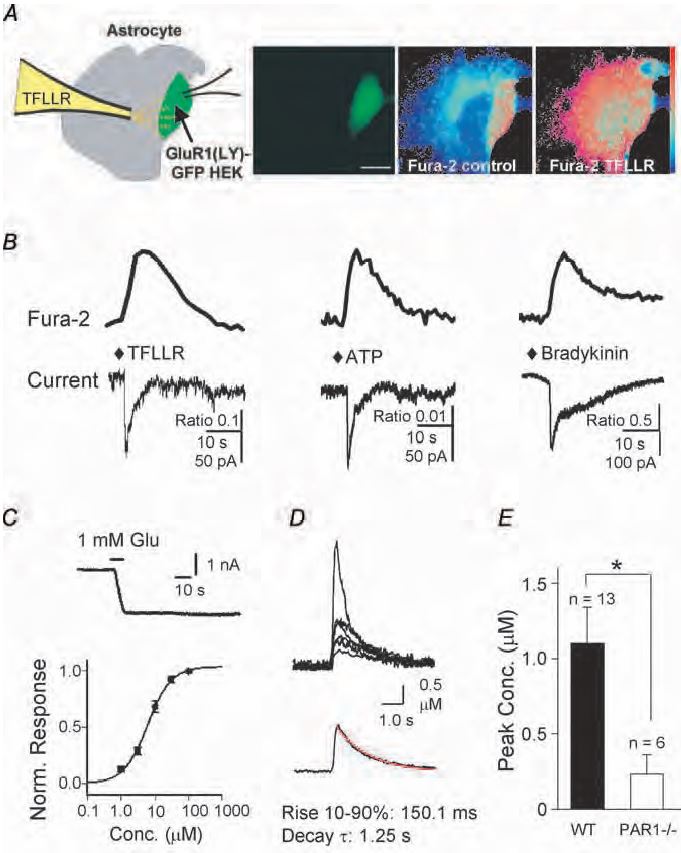

One important implication of our recent experiments (Lee et al. 2007) is that the level of glutamate estimated to be released from astrocytes (~1-6 µM; Fig. 1C, D, E) following activation of G-protein-coupled protease activated receptors is much lower than that in conventional neuronal synapses. Intra-cleft glutamate has been estimated to reach 1 mM during vesicular release (Clements et al. 1992), nearly a thousand times higher than the levels we estimate for astrocytic glutamate release. Even if our estimation of glutamate release was off by several fold, the extracellular concentration still would be sufficient to activate NMDA receptors, but insufficient to activate AMPA receptors. Another feature of our results is that we find different G-protein coupled receptors with similar whole cell Ca2+ signalling (Fig 2) mediate different levels of glutamate release. For example, activation of P2Y G-protein-coupled receptors release enough glutamate (estimated peak of 6 µM) to cause significant activation of group I metabotropic glutamate receptors (EC50 10 µM), which can potentiate NMDA receptor responses (Wirkner et al. 2007). By contrast, the release of 1 µM glutamate by protease-activated receptor-1 is predicted to cause little mGluR1 activation, even though this level can trigger localized postsynaptic depolarizations. Thus, there may be mechanisms that render compart-mentalization and specificity for astrocytic G-protein coupled receptor signalling to neurones.
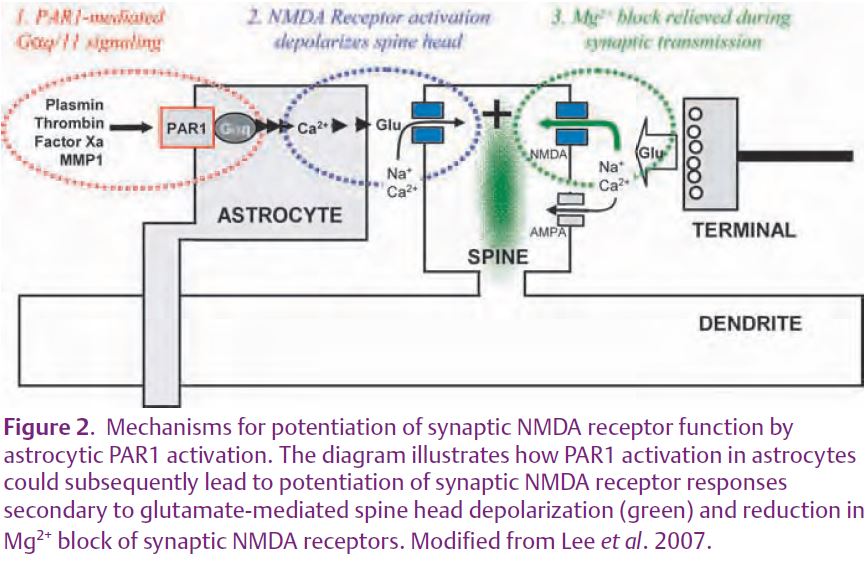
In the last decade a watershed event has been the acceptance that astrocyte-neuronal communication can take place by many mechanisms, including glutamate release from astrocytes. Our data suggest that this can provide a unique mechanism of NMDA receptor potentiation via relief of Mg2+ block. This mechanism may reflect spine head depolarization and consequent reduction of NMDA receptor Mg2+ block during subsequent synaptic currents (Fig. 2). Our study provides compelling evidence that astrocytes not only remove synaptically released glutamate, but also actively release glutamate in a Ca2+-dependent fashion to shape the synaptic NMDA responses at nearby synapses. Furthermore, the mechanisms described here are most likely to be shared by a wide range of astrocytic receptors that can elevate intracellular Ca2+. Perhaps most importantly, mechanisms within astrocytes could be potential targets for new therapies to alleviate cognitive and muscle-control deficits for people with brain or spinal cord damage. Thus, true to its form, a star-shaped brain cell may finally be ready to play a starring role in modulation of synaptic transmission.
References
Araque A, Li N, Doyle RT & Haydon PG (2000). SNARE protein-dependent glutamate release from astrocytes. J Neurosci 15, 666–673.
Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T & Volterra A (1998). Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391, 281–285.
Clements JD, Lester RA, Tong G, Jahr CE & Westbrook GL (1992). The time course of glutamate in the synaptic cleft. Science 258, 1498–1501.
Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J & McCarthy KD (2007). Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626.
Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB & Traynelis SF (2007). Astrocytic control of synaptic NMDA receptors. J Physiol 581,1057–1081.
Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S & Haydon PG (1994). Glutamate-mediated astrocyte-neuron signalling. Nature 369, 744–747.
Wirkner K, Gunther A, Weber M, Guzman SJ, Krause T, Fuchs J, Koles L, Norenberg W & Illes P (2007). Modulation of NMDA receptor current in layer V pyramidal neurons of the rat prefrontal cortex by P2Y receptor activation. Cereb Cortex 17, 621–631.