
Physiology News Magazine

Airway liquid secretion and cystic fibrosis lung disease
Stephen Ballard describes recent studies into anion and liquid secretion in airways and discusses how inhibition of this process provides important insights into the pathogenesis of CF
Features
Airway liquid secretion and cystic fibrosis lung disease
Stephen Ballard describes recent studies into anion and liquid secretion in airways and discusses how inhibition of this process provides important insights into the pathogenesis of CF
Features
Stephen T. Ballard
Department of Physiology, College of Medicine, University of South Alabama, USA
https://doi.org/10.36866/pn.54.22

Persons afflicted with cystic fibrosis (CF) suffer from numerous maladies including pancreatic insufficiency, intestinal obstruction, and sterility (in males). However, it is the pulmonary complications of the disease that cause premature death and morbidity in the vast majority of cases.
CF lung disease is characterized by the production of a thickened airway mucus, impairment of mucociliary transport, unusually high susceptibility to microbial infection of the airways, and gradual degeneration of the lung due to a constant state of inflammation.
CF is caused by mutations in the gene that codes for the CFTR, a transmembrane protein that normally functions as a cAMP-regulated chloride channel. In the lung, the CFTR resides in the apical membranes of the serous cells of submucosal glands and the epithelial cells that line the airway surfaces.
Our laboratory focuses on the possible role that CFTR-mediated chloride and liquid secretion plays in the normal lung and how disruption of this process might lead to the pulmonary pathology seen in CF.
To better understand why chloride secretion might be important to normal airway function, we first looked at different airway regions to see if this process was localised in the lung. In the distal airways of the pig lung, whose morphology closely resembles the human lung, the basal rates of chloride secretion are far greater in the bronchi, the muscular thick-walled airways that express many submucosal glands, than in the bronchioles, the thin-wall compliant airways that are aglandular (Ballard et al. 1995). When the bronchi are exposed to glandular secretogogues, such as acetylcholine or substance P, the glands respond by vigorously secreting not only chloride but also bicarbonate which together drive the secretion of liquid by these tissues (Ballard et al. 1999, Trout et al. 2001).
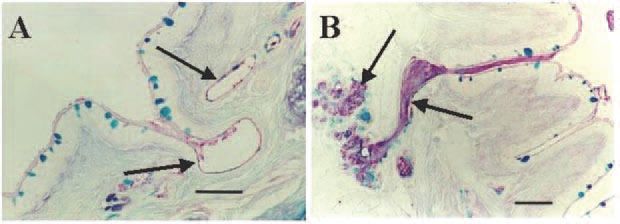
Normally, when submucosal glands of the airways are stimulated, they secrete not only liquid from the serous cells but also gel-forming mucins from mucous cells. We reasoned that, if we selectively inhibited the liquid component of gland secretion and stimulated the glands to secrete, then mucin and fluid secretion would become uncoupled, leading to the secretion of a very thick, low-volume mucus. Indeed, when we pretreat pig bronchi with bumetanide, a loop diuretic that inhibits chloride secretion, and dimethylamiloride (DMA), a sodium-proton exchange inhibitor that blocks bicarbonate secretion, and then stimulate the airways with acetylcholine, the glands become impacted with mucin, replicating the earliest sign of CF airway disease (Fig. 1) (Inglis et al. 1998). The same anion secretion inhibitors also lead to the production of a thickened airway mucus with altered rheological properties; however, these changes alone are probably not adequate to impair mucociliary transport (Trout et al. 1998).
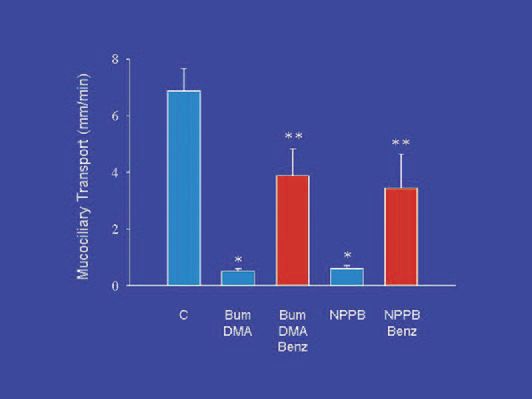
Airway surface liquid (ASL) normally exists in two phases: a low viscosity ‘sol’ phase, which occupies the periciliary space, and a high viscosity ‘gel’ phase, which contains the gel-forming mucins and is usually excluded from the periciliary space. For mucociliary transport to function normally, the cilia of the surface epithelial cells must beat within the sol phase which is maintained by a balance of liquid secretion (originating from glands and probably from surface epithelium as well) and liquid absorption (driven by active sodium absorption across the surface epithelium). When blockers of chloride and bicarbonate secretion are used to disable liquid secretion in porcine tracheas in vitro mucociliary transport is nearly abolished (Ballard et al. 2002)(Fig. 2). However, if the tissues are also exposed to benzamil, an inhibitor of epithelial sodium channels (ENaC) which mediate absorption, mucociliary transport is preserved at approximately one-half the control rate. When bumetanide and DMA are infused into an isolated, vascular perfused pig lung to block airway liquid secretion, the periciliary fluid becomes depleted, and the cilia are flattened between the dense mucus gel layer and the apices of the epithelial cells (Trout et al. 2003) (Fig. 3). Therefore, the impairment of mucociliary transport is likely to be due to the depletion of airway surface liquid – a consequence of reduced liquid secretion in the face of ongoing liquid absorption.
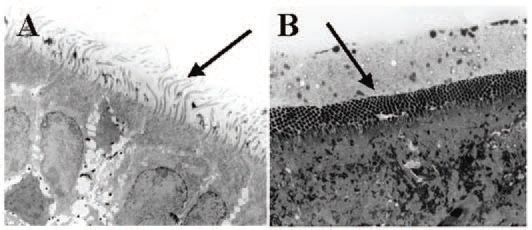
These findings provide important insights into the aetiology of CF airway disease. If the secretion of liquid by the glandular airways is critically dependent upon CFTR, it is obvious that these same problems would occur in CF bronchi. Indeed, CF is characterized by mucin occlusion of submucosal glands, production of thickened mucus, and impaired mucociliary transport. One expects that inhaled bacteria, which become embedded in the airway surface mucus, would not be easily cleared because of the reduced mucociliary transport, predisposing the lungs to colonization. A possible caveat to this hypothesis, however, comes from a recent study suggesting that liquid secretion by submucosal glands is only partially disrupted in CF (Joo et al. 2002). A milder disruption in liquid secretion by glands might therefore take longer to manifest into the severe impairments of mucociliary transport that we see in the pig model and possibly explain why it may take several years for severe lung disease to develop in CF patients. Clearly, this issue will be the critical focus of future research efforts.
References
Ballard ST, Fountain JD, Inglis SK, Corboz MR & Taylor AE (1995). Chloride secretion across distal airway epithelium: relationship to submucosal gland distribution. Am J Physiol 268, L526-L531.
Ballard ST, Trout L, Bebök Z, Sorscher EJ & Crews A (1999). CFTR involvement in chloride, bicarbonate and liquid secretion by airway submucosal glands. Am J Physiol 277, L694-L699.
Ballard ST, Trout L, Mehta A & Inglis SK (2002). Liquid secretion inhibitors reduce mucociliary transport in glandular airways. Am J Physiol 283, L329-L335.
Inglis SK, Corboz MR,& Ballard ST (1998). Effect of anion secretion inhibitors on mucus content of airway submucosal glands. Am J Physiol 274, L762-L766.
Joo NS, Irokawa T, Wu JV, Robbins RC, Whyte RI & Wine JJ (2002). Absent secretion to vasoactive intestinal peptide in cystic fibrosis airway glands. J Biol Chem 277, 50710-50715.
Trout L, Corboz MR & Ballard ST (2001). Mechanism of substance P-induced liquid secretion across porcine bronchial epithelium. Am J Physiol 281, L639-L645.
Trout L, King M, Feng W, Inglis SK & Ballard ST (1998). Inhibition of airway liquid secretion and its effects on the physical properties of airway mucus. Am J Physiol 274, L258-L263.
Trout L, Townsley MI, Bowden A & Ballard ST (2003). Disruptive effects of anion secretion inhibitors on airway mucus morphology in isolated perfused pig lungs. J Physiol 549, 845-853.