
Physiology News Magazine

Ca2+ sparks – SOS signals of struggling muscle
Ca2+ sparks are rare in mammalian skeletal muscle under physiological conditions. Our recent studies reveal an important role for mitochondria in suppression of these localized Ca2+ signals
Features
Ca2+ sparks – SOS signals of struggling muscle
Ca2+ sparks are rare in mammalian skeletal muscle under physiological conditions. Our recent studies reveal an important role for mitochondria in suppression of these localized Ca2+ signals
Features
Elena V Isaeva, Vyacheslav M Shkryl, & Natalia Shirokova
Department of Pharmacology and Physiology, University of Medicine and Dentistry of New Jersey, New Jersey Medical School, Newark, NJ, USA
https://doi.org/10.36866/pn.62.27

The primary function of skeletal muscle is to produce shortening and force for movement of our bodies. A complex series of events, known as excitationcontraction coupling (ECC), links nerve-mediated electrical excitation of the skeletal muscle membrane to muscle contraction. A key step in ECC is the release of Ca2+ from intracellular Ca2+ stores (sarcoplasmic reticulum, SR) to activate the contractile machinery. Ca2+ release occurs through specialized Ca2+ channels, called ryanodine receptors (Ryrs). For several decades it was believed that Ca2+ release channels are opened by mechanical interactions with the voltage-sensing molecules of the Ttubular membrane (dihydropyridine receptors) and that Ca2+ released from the SR by this voltage-dependent mechanism subsequently activates additional Ryrs via Ca2+-induced Ca2+ release (CICR), thereby amplifying the contractile signal.

The phenomenon of CICR was discovered in mechanically skinned fibres in the early1970s (Endo et al. 1970) but for a long time there was no direct evidence of its involvement in Ca2+ signaling during ECC.
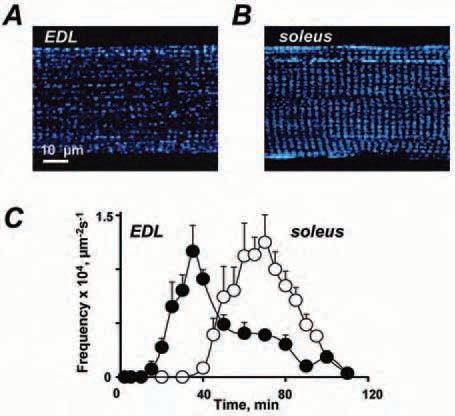
Direct experimental evidence for CICR function in normal skeletal muscle physiology was provided only in 1995, when Ca2+ sparks were found in amphibian skeletal muscle. Sparks, transient localized elevations of cytosolic [Ca2+], represent SR Ca2+ release events through a small cluster of Ryrs, and are considered to be elementary events of CICR. They are abundant in frog muscle fibres and their frequency is greatly increased by electrical stimulation, suggesting a substantial contribution of CICR to ECC. Was the missing link of ECC definitively found? No! In mammalian muscle, spontaneous Ca2+ sparks are rarely seen and depolarization does not induce Ca2+ sparks, but Ca2+ ‘embers’, which probably result from the opening of a single Ryr channel and do not involve CICR. The latter observations left us with burning questions: why are there no Ca2+ sparks in mammalian muscle? Is there a fundamental difference in Ca2+ signalling between mammals and amphibians?
In the search for answers, several hypotheses have been considered, but most turned out to be dead-end roads. Initially, it had been suggested that the expression of the Ryr3 isoform might be necessary for skeletal muscle to produce Ca2+ sparks, because most amphibian, but not adult mammalian, muscles express Ryr3. However, this hypothesis did not survive experimental challenge as spontaneous Ca2+ sparks were found in developing skeletal muscle of Ryr3-knockout mice. Alternatively, in Shirokova et al. (1999), we proposed that a functional interaction between DHPrs and Ryrs prevents CICR. But this idea was subsequently challenged by Kirsch et al. (2001), who demonstrated abundant sparks in mechanically skinned mammalian fibres, where the coupling between DHPrs and Ryrs is presumably preserved. This indicated that mammalian fibres are capable of producing Ca2+ sparks, but these are normally suppressed, and immediately raised another question: why would skinning of the cells lead to the appearance of Ca2+ sparks?
Our recent studies revealed an important role of mitochondria in the suppression of sparks in mammals. For a long time, the almost exclusive function of mitochondria in skeletal muscle was considered to be production of the energy (ATP) supply necessary for the key muscle function – contraction. However, increasing evidence indicates that mitochondria exert a multifactorial influence on cell physiology: the organelles can accumulate Ca2+, produce reactive oxygen species (ROS), balance cytosolic redox potential, etc. Each of these functions is influenced by, and can in turn influence, intracellular Ca2+ homeostasis. Importantly for the interactions within these Ca2+ dependent feed-back loops, in mammalian muscle, mitochondria are strategically located close to the SR Ca2+ release sites, where they are capable of interfering with and sensing SR Ca2+ release (Shkryl & Shirokova, 2005).
How can mitochondria suppress CICR? In Isaeva & Shirokova (2003) we first suggested that a degradation of mitochondrial function following muscle permeabilization and consequent washout of cytosolic constituents is a proximate cause for the appearance of Ca2+ sparks in permeabilized mammalian fibers. Experiments with several mitochondrial uncouplers and Ru360, a specific inhibitor of mitochondrial Ca2+ uptake, revealed a potential role of mitochondrial Ca2+ buffering in the inhibition of CICR, as impairment of mitochondrial Ca2+ uptake boosted spark activity. In search of other mechanisms for mitochondria to affect cytosolic Ca2+ signals we next studied the appearance of sparks in permeabilized skeletal muscle fibres with different mitochondrial content (Isaeva et al. 2005). The results were striking: in all fibre types, Ca2+ sparks developed in parallel with the decay of the mitochondrial redox potential.
Moreover, the appearance of sparks was delayed or sped up by the addition of exogenous ROS scavengers or ROS generators, respectively. These findings strongly implied that Ca2+ sparks occur when ROS increase in the cytosol as the redox potential falls upon impairment of mitochondrial metabolism. In agreement with this view, sparks were reported in intact mammalian muscle cells in response to osmotic stress or strenuous exercise (Wang et al. 2005), conditions in which the cytosolic ROS balance is likely to be compromised. Interestingly, it also turned out that stress-induced Ca2+ spark activity was significantly augmented in muscle fibers dissected from mdx mice lacking dystrophin, a widely used model of muscle dystrophy. This last observation suggests that Ca2+ sparks, although not the mediator of Ca2+ signaling in normal ECC of mammals, may play an important role in muscle pathology.
In summary, it is clear that CICR is less important for mammalian skeletal muscle ECC under physiological conditions than for amphibian muscle. Mammalian fibres have two triadic junctions per sarcomere whereas lower animals have only one. Ca2+ released from the SR in mammals thus needs to travel a shorter distance to activate the contractile machinery of the muscle. Therefore, a further amplification of the initial voltage-induced Ca2+ signals by CICR may not be necessary. Under stress, however, a moderate augmentation of cytosolic Ca2+ signals may be required for muscle adaptation.
In particular, increased cytosolic Ca2+ concentration could lead to a mild increase of the mitochondrial Ca2+ load. This, in turn, can counteract the decreased mitochondrial metabolism by stimulating ATP synthesis, thereby improving muscle contractility. Under pathophysiological conditions associated with mitochondrial dysfunctions, acute and exaggerated Ca2+ spark activity could be a precursor of muscle degeneration.
It still remains to be clarified exactly which mitochondrial mechanisms normally suppress CICR in mammals, to what extent this suppression can be impaired in muscle diseases, and how a failure of this suppression is involved in muscle pathophysiology. It may turn out that some of these mechanisms could be targets for pharmacological interventions to slow down, or prevent, the progression of degenerative muscular conditions.
Acknowledgements
Our work was supported by the NIH and MDA.
References
Endo M, Tanaka M & Ogawa Y (1970). Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle. Nature 228, 34-36.
Isaeva EV & Shirokova N (2003). Metabolic regulation of Ca2+ release in permeabilized mammalian skeletal muscle fibres. J Physiol 547, 453-462.
Isaeva EV, Shkryl VM & Shirokova N (2005). Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J Physiol 65, 855-872.
Kirsch WG, Uttenweiler D & Fink RH (2001). Spark- and ember-like elementary Ca2+ release events in skinned fibres of adult mammalian skeletal muscle. J Physiol 537, 379-389.
Shkryl VM & Shirokova N (2005). Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. J Biol Chem Oct 10; [Epub ahead of print]
Shirokova N, Shirokov R, Rossi D, González A, Kirsch WG, García J, Sorrentino V & Ríos E (1999). Spatially segregated control of Ca2+ release in developing skeletal muscle. J Physiol 521, 483-495.
Wang X, Weisleder N, Collet C, Zhou J, Chu Y, Hirata Y, Zhao X, Pan Z, Brotto M, Cheng H & Ma J (2005). Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat Cell Biol 7, 525-530.