
Physiology News Magazine

Developing light sheet microscopy in an advanced imaging facility
Light Sheet Microscopy offers unprecedented temporal resolution for in toto imaging of live specimens. While the advantages of this relatively simple microscopic set up are self-evident, users should be mindful of the sheer quantity of data generated and careful planning is necessary.
Features
Developing light sheet microscopy in an advanced imaging facility
Light Sheet Microscopy offers unprecedented temporal resolution for in toto imaging of live specimens. While the advantages of this relatively simple microscopic set up are self-evident, users should be mindful of the sheer quantity of data generated and careful planning is necessary.
Features
Kevin O’Holleran
University of Cambridge, UK
https://doi.org/10.36866/pn.95.40
At the Cambridge Advanced Imaging Centre (CAIC) we are building a complete portfolio of advanced fluorescence imaging, from two-photon to super-resolution and light sheet microscopes (LSMs). The mission of the centre is to make available the most advanced imaging techniques to a wide variety of life sciences by designing and developing the instruments in-house by an interdisciplinary team of physicists, engineers, computing scientists and biologists. Of all the techniques we are implementing, light sheet microscopy is our primary focus due to the incredible range of bioimaging applications it enables.
There has been a significant amount of excitement surrounding light sheet microscopy over the last few years. Although not a new technique, having appeared in various guises since the early 1900s, it has recently gone from strength to strength, enabling a broad range of biological imaging with unprecedented temporal resolution of live specimens, both in vivo and in toto. The primary advantage to light sheet microscopy is its combination of speed and low photodamaging effects, the latter being particularly essential for imaging of whole live organisms.
In recent years there has been an abundance of incredible imaging made possible by LSMs. Through use of multiple light sheets an entire larval zebrafish brain was imaged, via a genetically encoded calcium indicator, in 3D at a speed of 0.8 Hz continuously for an hour whilst capturing 80% of all neurons at a single-cell resolution (Ahrens et al. 2013). This fast and long term imaging combined with multiple sheets also makes cell lineage tracking possible in full Drosophila embryo development, by imaging in 3D once every 20 s over a number of hours (Krzic et al. 2012). Speed, in the case of imaging Drosophila, is essential given how often and quickly mitotic divisions occur, taking up to only a minute to complete. An example of a Drosophila embryo imaged with our LSM (see Fig. 2C). LSMs equipped with high NA excitation and observation lenses have also enabled fast 3D imaging of mitosis in single cells, with each volume measurement taking only one second (Planchon et al. 2011).
In order to operate over a wide variety of specimens and applications, LSMs come in many different configurations, but the principle with which they all work is the same. At this point I will discuss briefly the general principles of how light sheet microscopes work and some common issues with their application but I will keep the technical details brief and concentrate on the practicalities, benefits and impact of using LSMs in an imaging facility. For a fuller review of different types of LSM configurations and their use in developmental biology see Huisken & Stainier (2009).
The mechanism through which LSMs achieve fast, low bleaching, imaging is through the illumination of a fluorescent sample from a direction orthogonal to the detection objective lens. See Fig. 1 for an illustration of such a geometry compared with a confocal system. This illumination forms a sheet of light which excites only a thin slice of the sample. The excited fluorescent markers emit light and the detection objective images the fluorescence onto a large multi-megapixel camera chip. The result is an optically sectioned image, captured with a single exposure of laser light formed into a sheet and imaged with a camera exposure typically of tens of milliseconds.
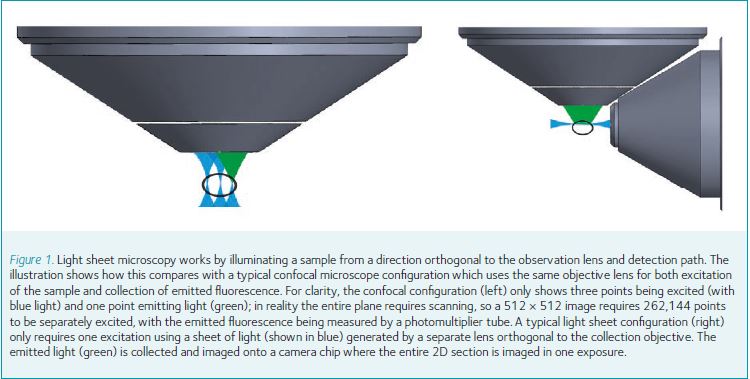
Compare this with a laser scanning confocal microscope: a 1000 × 1000 image requires a million measurements, each measurement requiring the sharp focus of laser light for several microseconds, with sectioning achieved by the rejection of out-of-focus light. This comparison is a little over-simplified but it demonstrates clearly the principle advantages over laser scanning confocal systems.
Like all techniques, LSMs have their disadvantages. A significant one is non-uniformity of image properties caused by non-uniformity of the light sheet used. A common non-uniformity is the varying thickness, and intensity, of the light sheet across the direction of propagation. Light sheets are thinner, and brighter, at the focal plane and gradually get thicker either side of the focus. Exciting a sample with such a light sheet results in an optically sectioned image with varying section thickness, making the edges of the image appear more blurred than the centre. Other non-uniformities are caused by variations in the structure of the sample being excited. As a light sheet propagates in a sample, it can be absorbed and scattered, leading to artefacts such as shadows being cast across the image as streaks and further thickening of the light sheet. These problems can be overcome to some extent by using different types of beams, such as Bessel beams (Fahrbach et al. 2010), which have a longer depth of field than normal Gaussian beams; by using multiple light sheets incident from different directions (Huisken & Stainier, 2007); by using two-photon excitation (Truong et al. 2011) for deeper penetration and less scattering; or even by using a combination of the above. All these approaches can indeed both improve image quality deeper into the sample (as seen from the excitation lens) and provide more isotropic image resolution, but at a cost of higher exposure to laser light, albeit still orders of magnitude less than confocal laser scanning systems. However, these improvements come at a cost of increased instrument complexity and expense.
Putting the more advanced variants of LSMs aside, the relative simplicity of a LSM to other systems has made it popular as a DIY microscope, as evidenced by the openSPIM project (Pitrone et al. 2013) which details exactly how to build your own selective plane illumination microscope (SPIM), a type of light sheet microscope that utilises a cylindrical lens to form a fixed light sheet with samples mounted vertically in a rotatable holder (Huisken et al. 2004). The openSPIM project lists what parts to use and how to put them together, and even provides control software through the Micromanager platform. A typical openSPIM experiment requires samples to be suspended in an agarose cylinder which can then be immersed in water (or other similar refractive index medium) and moved and rotated relative to horizontally mounted lenses and a vertical fixed light sheet. Although suitable for Drosophila and zebrafish embryos it is not suitable for samples that cannot be suspended in agarose such as chick or mouse embryos. Deviating from the openSPIM design does, however, require a reasonable knowledge of optics and the hardware and software platform being used for controlling the system.
At CAIC we are building different types of LSMs, designed by ourselves and our collaborators. The first system we have built is aimed at imaging a wide range of model organisms and as such was built to allow flexible sample mounting. The objective lenses are mounted at 45 degrees from the vertical and samples are simply mounted on a bar in a Petri dish (the configuration can be seen in Fig. 2A). This only allows illumination from one direction and as such we will soon be including a two-photon imaging mode to allow deeper penetration of the light sheet into the sample and removal of some shadowing effects.
Although the speed and low bleaching offered by LSMs enable a wide range of exciting biological imaging investigations it does leave the user with one big headache: enormous amounts of data. For example, the LSM we have built at CAIC can generate just over 800 Mb per second at its fastest (full frame at 100 fps or smaller frames even faster) and although different applications require different numbers of time points, colour channels and duration, we find that typical experiments result in several terabytes of raw image data. For example, imaging the retina of a 1.5 day post-fertilisation zebrafish embryo (Fig. 2B) for 48 h imaging 532 μm × 532 μm × 100 μm at a resolution of 0.26 μm × 0.26 μm × 0.5 μm every 2 min amounts to approximately 2 terabytes per channel. It won’t be uncommon for multi-channel LSM data sets to reach 5 Tb of data and many hundreds of thousands of images, particularly for developmental biology applications.

References
Ahrens MB, Orger MB, Robson DN, Li JM & Keller PJ (2013). Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat Methods 10, 413–420.
Fahrbach FO, Simon P & Rohrbach A (2010). Microscopy with self-reconstructing beams. Nat Photonics 4, 780–785.
Huisken J & Stainier DYR (2007). Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM). Opt Lett 32, 2608–2618.
Huisken J & Stainier DYR (2009). Selective plane illumination microscopy techniques in developmental biology. Development 135, 1963–1975.
Huisken J, Swoger J, Bene F, Wittbrodt J & Stelzer EHK (2004). Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 305, 1007–1009.
Krzic U, Gunther S, Saunders TE, Streichan SJ & Hufnagel L (2012). Multiview light-sheet microscope for rapid in toto imaging. Nat Methods 9, 730–733.
Pitrone PG, Schindelin J, Stuyvenberg L, Preibisch S, Weber M, Eliceiri KW, Huisken J & Tomancak P (2013). OpenSPIM: an open access light sheet microscopy platform. Nat Methods 10, 598–599.
Planchon TA, Gao L, Milkie DE, Davidson MW, Galbraith JA, Galbraith CG & Betzig E (2011). Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat Methods 8, 417–423.
Truong TV, Supatto W, Koos DS, Choi JM & Fraser SE (2011). Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat Methods 8, 757–760.