
Physiology News Magazine

Distinguishing acute and chronic effects of placental dysfunction on maternal blood pressure
Features
Distinguishing acute and chronic effects of placental dysfunction on maternal blood pressure
Features
Annemarie Hennessy (1) & Angela Makris (2)
1: School of Medicine, University of Western Sydney and Director of Medicine, Campbelltown Hospital
2: Renal Unit, Liverpool Hospital, University of New South Wales, Sydney Australia
https://doi.org/10.36866/pn.72.33

Pregnancy is a state of adaptive hypotension. The mechanism of this hypotension is still poorly understood but consists of a dramatic endocrinological reaction to placental vasodilators and low grade immune reactive responses.
In pregnancy, the links between cardiac output, systemic vascular resistance and blood pressure were finally determined by physiological studies performed in primates (Phippard et al. 1986) and later confirmed in humans (Clapp et al.1988). The prior view was that the decrease in blood pressure was due to changes in cardiac status. The primary change is in fact a decrease in vascular resistance (pulmonary and systemic) which leads to a compensatory renin aldosterone angiotensin system (RAAS) activation to restore blood volume and mitigate against any further fall in maternal blood pressure. Resistance to normal pressor responses is also a hallmark of pregnancy (Gant, 1973) and identifies the profound nature of the vasodilatory response. Recent findings of angiotensin 1/bradykinin receptor role in this resistance are intriguing (Granger, 2006).
Pre-eclampsia is one of the commonest medical complications of pregnancy and accounts for 75000 maternal deaths per year and 300 000 perinatal deaths. The hallmark of this disease is hypertension and the accompanying cerebral, hepatic and renal complications demonstrate a widespread endothelial injury. This is best typified by endothelial swelling and disruption in the renal glomerulus which presents clinically as proteinuria. The increase in maternal blood pressure is due to an increase in systemic vascular resistance which is profound and demonstrates a reversal of the changes outlined above, i.e. a loss of pressor resistance, a decline in RAAS activation and a decline in cardiac output. The changes in cardiac function are also due to endocardial dysfunction as part of this widespread abnormality (Simmons, 1999), and happen surprisingly quickly.

A great discovery with regard to preeclampsia over the last 4 years has been the identification of endothelial cell ‘toxins’ which are biologically plausible as mediators of endothelial dysfunction (Fig. 1). These include angiogenic factor fms-like-tyrosine kinase-1 (sFlt-1) which antagonizes the vasodilator vascular endothelial growth factor (VEGF), and placental growth factor (PlGF). Also, the immune modifier endoglin is a circulating factor in preeclampsia. In the past, placental function tests (e.g. oestrial concentrations), and tests of maternal endothelial function (fibronectin) have been abandoned as unhelpful in diagnosis and unsuitable targets for therapy due to their non-specific nature. These newly recognized circulating factors have specific actions which imply they link endothelial dys-function with placental dysfunction. sFlt-1 links the formation of the placental vasculature and its hypoxia responses to maternal vascular resistance, and endoglin, to the early immunological events of pregnancy.
The pathological phases of pre-eclampsia can be described as pre-clinical and clinical. In the preclinical phase, the aberrant formation or placental invasion of maternal blood vessels occurs. In the clinical phase, the mother responds via endothelial damage with hypertension and proteinuria. The early placental events are immunological and vascular in origin. The result, therefore, is the active inhibition of robust maternal immunity and the invasion of uterine (maternal) spiral arteries to form a low pressure high flow system without local autoregulation.
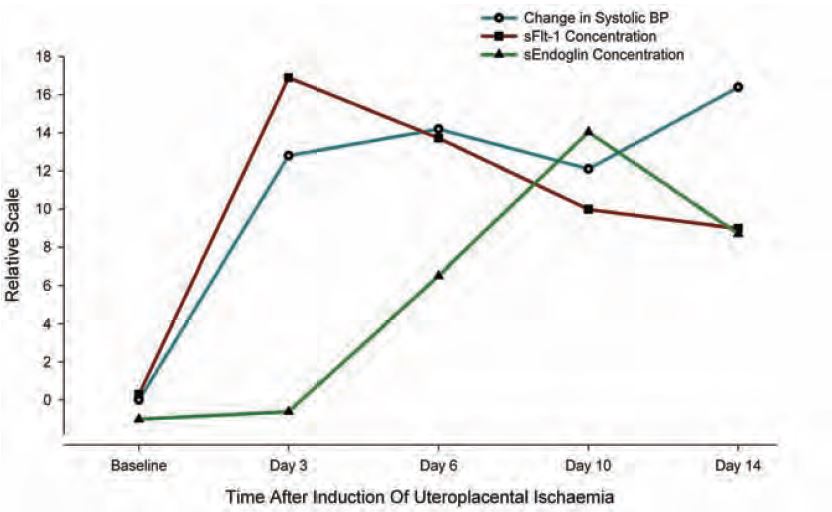
Soluble Flt-1 is the circulating form of the VEGF receptor Flt-1. Binding of VEGF and Flt-1 is responsible for angiogenesis and blood vessel integrity. The central role of sFlt-1 as a mediator of pre-eclampsia is based on infusion studies in pregnant rats (Maynard, 2004). The animals studied subsequently developed renal endothelial lesions and hypertension which were reversible with exogenous VEGF (Li et al. 2007). A cause of the elevation in sFlt-1 has been shown by us to be acute placental ischaemia (Fig. 2). This was confirmed recently in rats (Grainger, 2007). Studies of sFlt-1 in banked human serum has shown that elevations occur 3–8 weeks prior to the onset of clinical symptoms (Levine, 2004) suggesting that placental ischaemia later in pregnancy is the probable underlying cause.
Endoglin is the soluble receptor for TGFβ1-3. Receptor activation activates a predominantly immunosuppressive response. In pregnant rat studies, the addition of exogenous soluble endoglin leads to a more generalized endothelial disease than is seen with sFlt-1 alone (Venkatesa, 2006). The cause of any endogenous increase in endoglin is not known, but our data (Fig. 2) would suggest that it is a delayed response to reduced placental blood flow. Studies of human serum suggest that it is elevated up to 3 months prior to the clinical onset of hypertension (Levine, 2006), suggesting either that it is a more subtle and sustained marker of ischaemia much earlier in pregnancy or that the ischaemia is linked to an inflammatory response which builds more slowly. The presence of an early inflammatory aberration being linked with pre-eclampsia is evidenced by many studies of cytokine (Th1/Th2) imbalance and whole animal physiology studies showing that cytokine imbalance in early pregnancy can control maternal blood pressure (Orange, 2005).
It is not known why this immune imbalance occurs in early pregnancy. Whether this is due to innate overactivity of the maternal uterine natural killer cell or other T cells, or whether these cells are overactive as a result of poor immunosuppressor function by the placenta itself is yet to be determined. This immune regulation appears to be linked to the intrinsically low oxygen tension in the uterus. The high incidence of pre-eclampsia in women with intrinsically poor placental blood supply as occurs in chronic hypertension would suggest also that the blood flow is a primary phenomenon. The endothelial dysfunction that results from the up-regulation of molecules such as endoglin and sFlt-1 may be relevant in local uterine artery dysfunction as much as in the profound endothelial disease manifest by such a remarkable and sudden increase in blood pressure.
The incidence of immediate consequences of pre-eclampsia, headache, seizures, renal failure and liver failure are decreasing due to excellent antenatal care, accurate diagnosis and prompt delivery. Support for younger and younger babies by advances in neonatal intensive care have also diminished perinatal loss due to prematurity. The longer consequences to maternal and neonatal cardiovascular health after hypertension in pregnancy are clear to clinicians but as yet not fully understood. However, it is possible that the treatment and timing of delivery and therefore the duration of high blood pressure and endothelial dysfunction, may in and of themselves be contributors to the long term cardiovascular risk via endothelial health.
References
Capeless EL & Clapp JF (1989). Cardiovascular changes in early phase of pregnancy. Am J Obstet Gynecol 161,1449–1453.
Clapp JF 3rd, Seaward BL, Sleamaker RH & Hiser J (1988). Maternal physiologic adaptations to early human pregnancy. Am J Obstet Gynecol 159, 1456–1460.
Gant NF, Daley GL, Chand S, Whalley PJ & MacDonald PC (1973.) A study of angiotensin II pressor response throughout primigravid pregnancy. J Clin Invest 52, 2682–2689.
Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF et al. (2004). Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 350, 672–683.
Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, Epstein FH, Romero R, Thadhani R & Karumanchi SA (2006). CPEP Study Group. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 355, 992–1005.
Li Z, Zhang Y, Ying Ma J, Kapoun AM, Shao Q, Kerr I, Lam A, O’Young G, Sannajust F, Stathis P, Schreiner G, Karumanchi SA, Protter AA & Pollitt NS (2007). Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension 50, 686–692.
Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S et al. (2003). Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in reeclampsia. J Clin Invest 111, 649–658.
Orange S, Hennessy A, Thompson J, Rasko J, Vaughan J, Olive E, Pedlerm M & Horvath J (2005). Interleukin-10 regulates arterial pressure in early primate pregnancy. Cytokine 29,176–185.
Phippard AF, Horvath JS, Glynn EM, Garner MG, Fletcher PJ, Duggin GG & Tiller DJ (1986). Circulatory adaptation to pregnancy–serial studies of haemodynamics, blood volume, renin and aldosterone in the baboon (I). J Hypertens 4, 773–779.
Sholook MM, Gilbert JS, Sedeek MH, Huang M, Hester RL & Granger JP (2007). Systemic hemodynamic and regional blood flow changes in response to chronic reductions in uterine perfusion pressure in pregnant rats. Am J Physiol Heart Circ Physiol 293, H2080–H2084.
Simmons LA, Gillin AG & Jeremy RW (2002). Structural and functional changes in left ventricle during normotensive and preeclamptic pregnancy. Am J Physiol Heart Circ Physiol 283, H1627–H1633.
Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D ‘Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M & Karumanchi SA (2006). Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 126, 42–49.