
Physiology News Magazine

Films, muscle, potassium and exercise
David Jones recounts his quest, triggered by a 1970s movie scene, to find out why potassium is bad news for skeletal and cardiac muscle, yet good news for many other aspects of our physiology.
Features
Films, muscle, potassium and exercise
David Jones recounts his quest, triggered by a 1970s movie scene, to find out why potassium is bad news for skeletal and cardiac muscle, yet good news for many other aspects of our physiology.
Features
David Jones
Manchester Metropolitan University, UK
https://doi.org/10.36866/pn.90.26
Back in the 1970s I used to go to the cinema pretty frequently. One of the best of the actors of the time was George C Scott. Scott is better known as General Patton, in the film of that name. However, it is a little known film called The Hospital that is relevant here. The film is a very black comedy in which Scott plays Dr Bock, who has problems with his marriage, on top of which there is a series of murders of patients in his care. Things get so bad that Dr Bock is driven to contemplate suicide and there is one scene in which he is sitting with a tourniquet around his arm about to inject himself with a syringe full of potassium chloride. He doesn’t do it, and I can’t remember now what distracted him, possibly Dianna Rigg, his exceptionally attractive co-star, but I do remember sitting in the cinema thinking, “Why is KCl going to kill him?” After all, potassium is the major intracellular cation, it is found in nice things like fruit juices and bananas, and, surely drinking too much fruit juice isn’t going to kill you? By coincidence, this question, and the role of potassium in muscle and exercise physiology, was going to crop up at regular intervals over the next 40 years.
At that time I had just started working with Richard Edwards and David Hill at the Royal Postgraduate Medical Centre, Hammersmith Hospital, next door to Wormwood Scrubs prison. Richard had recently introduced the muscle biopsy technique into this country and he and David Hill were beginning to measure strength and function of human muscle, both of normal subjects and patients with a variety of muscle problems, and I was lucky enough to join them at the start. We were also very lucky that Brenda Bigland-Ritchie came to work with us while on a sabbatical year and we set about the most enjoyable part of any research project – just playing about and seeing what happens, but with a general interest in muscle fatigue.
One of the first things we noticed is that when stimulating the adductor pollicis muscle it is necessary to use a relatively high frequency of 80–100 Hz to obtain maximum force, but the muscle then rapidly fatigues. If a lower frequency is used, say 20 Hz, there is initially a lower force but the force is then maintained (Fig. 1). These experiments were carried out with the blood supply to the arm occluded with a tourniquet and the explanation seemed fairly obvious; with the high frequencies and high forces energy was being used up very rapidly, while with the low-frequency stimulation the energy reserves were lasting longer. However, as part of the general messing around, we tried changing the frequency during the high frequency stimulation at a time when the muscle was considerably fatigued and, to our surprise, reducing the stimulating frequency resulted in a rapid recovery of force. If the reason for the loss of force was the depletion of an energy reserve, say phosphocreatine, or the accumulation of something nasty, say lactic acid, then there would be no reason why simply reducing the stimulation frequency in an ischaemic muscle would result in an increase in force. So rather than this behaviour being explained by metabolic changes affecting the interaction of actin and myosin, it seemed more likely that some change in the electrical properties of the membrane was involved. At the time there was interest in the possibility of failure at the neuromuscular junction during fatiguing contractions and this might have been the explanation since we were stimulating via the motor nerve.
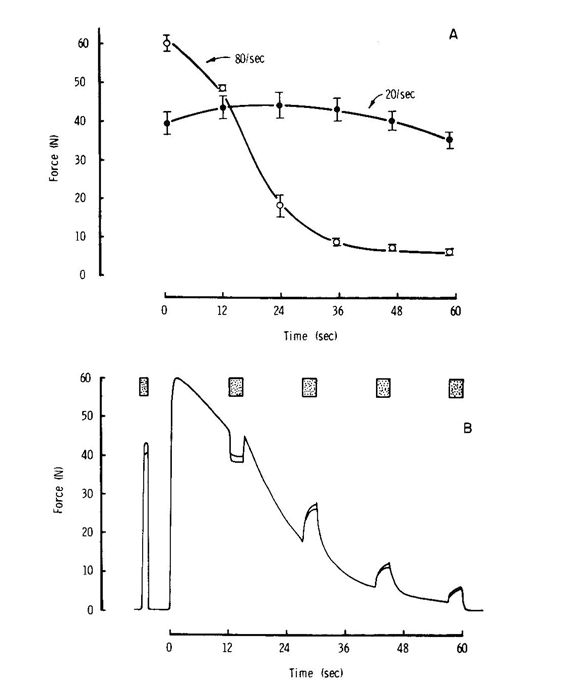
Transmission at the neuromuscular junction depends on the release of acetyl choline from synaptic vesicles and it is easy to imagine that when stimulating at high frequencies the rate of release outstrips the ability of the nerve ending to take up the choline, re-synthesise the acetyl choline and package it into new vesicles; reducing the frequency would thus give the nerve ending time to catch up. The test of this was to stimulate the muscle fibre membrane directly rather than through the nerve and neuromuscular junction, but this is difficult and painful to do with human muscle in situ. However, I had set up an isolated mouse muscle preparation where it was possible to block the neuromuscular junction with curare, which binds to the acetyl choline receptors, and stimulate the muscle directly with large plate electrodes. For no very good reason we did not believe the neuromuscular junction was the problem, so, to our delight, we found that this isolated, curarised, preparation behaved in exactly the same way as the human muscle, strongly suggesting that the problem lay with the electrical properties of the muscle fibres themselves, not the neuromuscular junction. This was further supported by EMG records from the human muscles where it was clearly seen that the action potential failed rapidly during high frequency stimulation and did not simply become smaller in amplitude, as would be expected if the neuromuscular junction was failing, but also became slower with an extended wave form (Fig. 2).
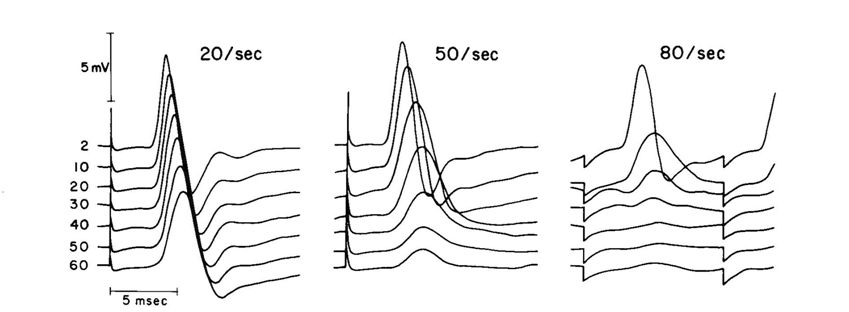
Skeletal muscle fibres are tightly packed together with little extracellular space and they also have a fantastic system of T-tubules which penetrate from the surface to the interior of the fibres. The surface area to volume ratio of these tubules is huge, so that movement of cations across the membrane can very rapidly change the extracellular concentrations. Our first thought was that since the action potential is due to opening of Na+ channels and an inward movement of sodium, then high frequency stimulation would deplete extracellular Na+ and thus the action potential would fail. The test of this was to artificially reduce the extracellular sodium, in which case we would expect the muscle to fatigue more rapidly; and, indeed, this is what happened (Fig. 3). So I was very pleased with my first bit of muscle research and even more pleased when after a struggle with the referees, who were supporters of the neuromuscular junction theory, the paper was published.
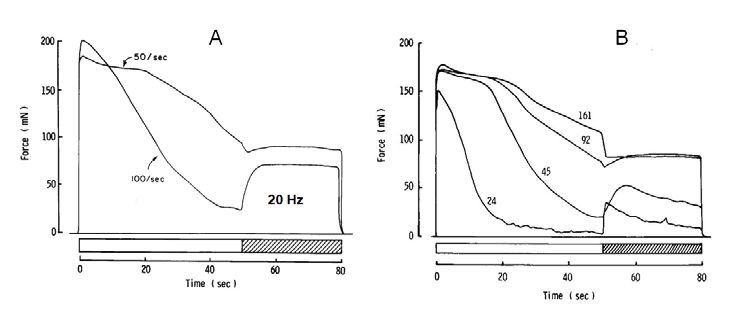
My excitement, however, was short lived, because it was obvious that the external sodium concentration had to be reduced a long way, down to a quarter of the normal level, before any substantial change in the rate of fatigue was observed. There was also the realisation that accumulation of potassium in the extracellular space could have a much more dramatic effect on muscle excitability. There had been a report from Paul Hník of considerable increases of extracellular K+ in working muscle, so, rather belatedly, we tried increasing the external K+ and found it to be very effective in reducing the excitability of muscle, not only reducing force but also producing a slowing of the action potential in a similar way to that seen with intact human muscle when stimulated at high frequency.
So now I had found out that a high external K+ is bad news for skeletal muscle and, in a similar way, I could see that it might also be bad news for cardiac muscle – something that Dr Bock already knew and, incidentally, was also known to those giving lethal injections where amongst the cocktail of drugs administered, potassium is the key lethal component. It is also known by cardiac surgeons who use a high-potassium saline solution to stop the heart beating during open heart surgery. What I wasn’t clear about is precisely why it is such bad news and I remember after one PhySoc meeting, where I had presented these results, it was pointed out that by raising the external potassium the resting membrane potential would come closer to the point where Na+ channels open and it would seem logical to expect that the muscle fibre would become easier to stimulate, not harder.
The sodium channel can be thought of as having two gates. One gate is normally shut when the internal resting potential is around -70 mV, but will open when the potential rises above a certain threshold and the sudden increase in Na+ permeability causes the interior of the fibre to become positive (the reversal potential). However, the second gate, which is normally open when the internal potential is negative, shuts as the interior of the fibre becomes positive so that the Na+ permeability is only high for a short interval before the membrane potential returns to the resting negative value as the K+ permeability again dominates the charge across the membrane. With the interior of the fibre again negative, the first Na+ gate shuts and the second gate opens ready for a next action potential. This all takes a matter of milliseconds and the closing of the Na+ channel is known as ‘fast inactivation’. However, things start to go wrong if there is an accumulation of external K+ and the resting membrane potential is raised (i.e. closer to zero) as, over a matter of several seconds, the Na+ channels start failing to open in the normal way in response to depolarisation, and this is known as ‘slow inactivation’. I had always thought the problem was due to temperamental behaviour of the second gate, but the evidence is that slow inactivation is not the fast inactivation mechanism going wrong, but is the result of as-yet poorly understood changes in the structure of the channel.
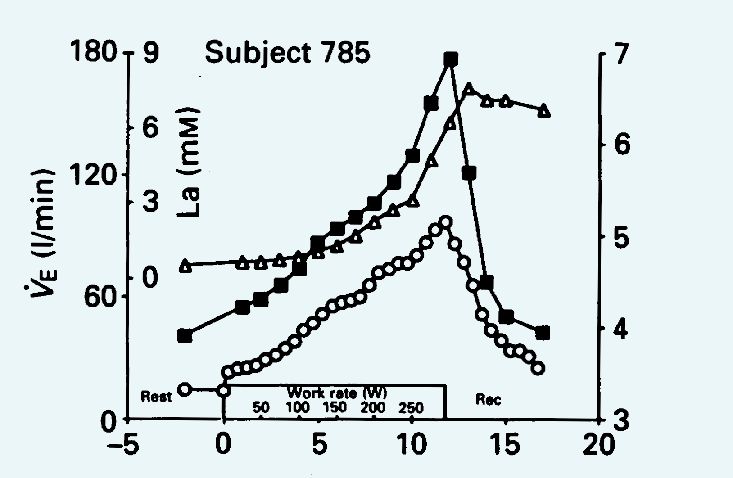
Whether or not the mechanism is fully understood, there is no doubt that high extracellular K+ is bad news for skeletal and cardiac muscle but the question is, how bad? Plasma potassium levels are normally tightly regulated between 3.5–5 mmol. Higher levels lead to concern and 7 mmol and above is described in the textbooks as constituting a medical emergency. Potassium is released from working muscle into the general circulation and there are many studies to choose from, but I like one in particular because it touches on a number of different aspects of the physiological role of potassium. In 1990, David Paterson and colleagues published a study concerned with the stimulus for increased breathing during exercise, in which they looked at the increase in ventilation in relation to the accumulation of potassium and lactate in the blood. It can be seen from Figure 5 that the K+ levels reach just about 7 mmol. David Paterson is the current Editor-in-Chief of The Journal of Physiology responsible for, amongst other things, the highest ethical standards of published work. Now, the subject in Figure 4 was clearly reaching dangerously high blood potassium levels and so I looked carefully through the paper to see how many subjects had died or what elaborate precautions had been taken to resuscitate them as they collapsed following a medical emergency, but I searched in vain. So does this paper fall below the normal high standards expected in The Journal?

Probably not – whilst these days there is increasing nervousness about doing almost anything with human subjects, in fact we all know that this type of exercise in healthy subjects is perfectly safe. But, given the fact that, as is widely reported in the literature, blood K+ levels can rise to 7 mmol or higher, it is not immediately clear why it should be safe.
Although 7 mmol K+ is a medical emergency for a subject lying in bed, the same K+ level is of little consequence with a subject exercising hard. The reason for this paradox lies in the action of the Na+/K+ transport ATPase which is stimulated by catecholamines released into the blood during exercise and by Na+ entering the active muscle fibres. The action of the transporter is two-fold. First, pumping K+ back into the cell minimises the accumulation of K+, and may be particularly important in the T-tubules where their small volume means that K+ concentrations can build up very rapidly. The second effect arises because the pump is asymmetric in that three Na+ are transported out of the cell for every two K+ coming in, exporting one more positive charge than enters the cell, acting as an electrogenic pump making the interior more negative than it would otherwise be. This counteracts the effect of potassium accumulating outside the fibre and thus the process of slow inactivation and is particularly important for the heart where raised catecholamines protect against the effects of high circulating K+ during exercise.
The Paterson paper clearly makes the point that K+, rather than acidosis as indicated by the increasing blood lactate, is the driving force for increased ventilation. The key point is in the recovery phase when the K+ concentration falls rapidly, but the lactate levels remain high for at least five minutes after the end of exercise and long after ventilation has returned to baseline. The paper also contains the results of exercise in McArdle’s patients who have the rare metabolic problem of not being able to breakdown glycogen in their working muscles, so they do not produce any lactate or become acidotic during exercise. The observation was that, as with the normal subjects, ventilation in the McArdle’s patients increased in parallel with the increased blood K+, but this was in the complete absence of any lactate.
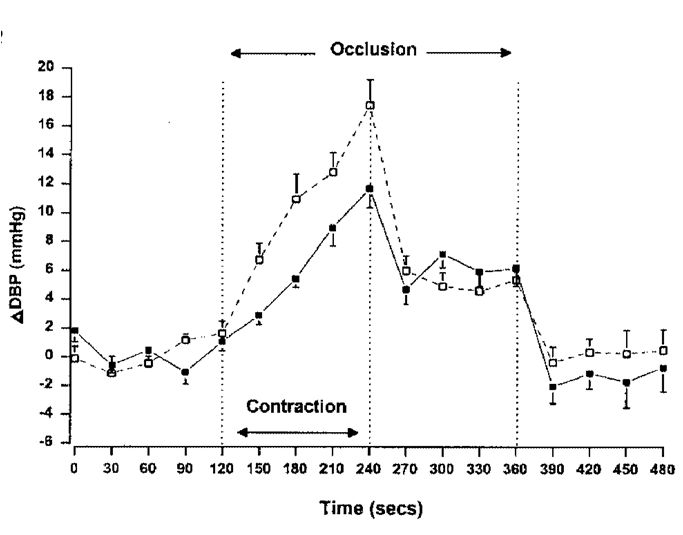
Potassium clearly has an important role to play by being at least one of the factors stimulating the drive to breathe harder during exercise. To get the oxygenated blood to where it is needed it is necessary to increase blood pressure and for the arterioles in the working muscles to vasodilate. There are a number of substances responsible for this vasodilation, but K+ released from the working muscles is an important component. Metabolites released from working muscles also influence blood pressure as has been demonstrated in some elegant experiments where a subject makes an isometric contraction with blood flow to the limb occluded with a pneumatic cuff. Blood pressure goes up during the contraction, but when the subject stops contracting the pressure falls to about half the peak value and stays there until the blood is allowed back when it rapidly returns to baseline (Fig. 5). The inference is that about half the rise in blood pressure during exercise is due to accumulating metabolites in the muscle, sometimes known as a ‘metaboreflex’.
There are a number of metabolites that could be responsible for this reflex, including both K+ and lactate, but the speed with which the blood pressure returns to baseline when the blood supply is restored, and the analogy with the Paterson study, suggest that K+ has a major role to play. The test of whether or not lactate is involved would be to study McArdle’s patients in this way, but the evidence is conflicting, with some reporting normal pressor responses, while others say it is reduced in these patients. It would, in any case, be both painful and dangerous, since these patients experience considerable pain and their muscles tend to go into contractures if they attempt prolonged contractions.
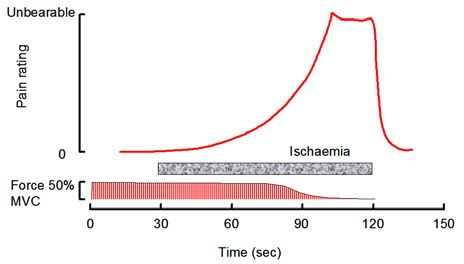
The mention of pain brings me to the final point about the role of potassium in muscle physiology. A very common form of muscle pain develops when running up a flight of stairs, lifting weights, riding a bicycle uphill or carrying heavy shopping. It has a very characteristic feel, often described as a burning sensation, and it is a warning that the muscles are coming to the limits of endurance. It is an easy form of pain to investigate experimentally and it was first studied by Sir Thomas Lewis, a cardiologist primarily interested in the pain of angina and claudication, who in the 1930s showed that the onset and extent of the pain varied with the frequency and strength of the contractions, so that pain is essentially proportional to the work done. Furthermore, if the muscle is held ischaemic the pain persists at much the same level after the contractions has stopped and does not go away until the blood returns. The pain disappears rapidly within 10–20 seconds and the feeling is very much of the pain being washed away as the blood returns (Fig. 6). Lewis concluded that the pain was due to the accumulation of some metabolite within the working muscles and the obvious question is whether the metabolite is K+ or lactate? Here the analogy with the Paterson study is very telling, partly because of the time course of events in the recovery period. The very rapid resolution of the pain is strikingly similar to the time course with which circulating K+ is cleared and very different to the slow clearance of lactate. But there is another parallel; if lactate is the cause of ischaemic pain then we would expect McArdle’s patients not to be affected, since they produce no lactate in their working muscles. In reality, severe pain during strong muscle contractions is a characteristic feature of these patients and, while it is impossible to tell if one person’s pain is the same as another, the patients describe the sensation in exactly the same way as everybody else.
So we see that an increase in extracellular potassium plays a number of roles. It can be potentially life threatening, reducing the excitability of skeletal and cardiac muscle, and it is responsible for the ischaemic pain signaling that the muscles are reaching the limit of performance. But it also helps by increasing ventilation, raising blood pressure and causing vasodilation all of which deliver oxygenated blood to the muscles that need it most. In fact, the release of K+ from working muscle may be seen as one of the ways of coordinating the responses of respiratory, cardiovascular and neuromuscular systems during exercise.
Chance and random events have a surprisingly large part to play in determining the direction of research just as they do for our careers and life in general. In my case, one such event took place in 1972 in a cinema at Clapham Common as I sat there wondering how it was that KCl was going to kill Dr Bock. It has taken a long time for me to realise the full implications of what potassium can do.
References
Bigland-Ritchie B, Jones DA & Woods JJ (1979). Excitation frequency and muscle fatigue: electrical responses during human voluntary and stimulated contractions. Exp Neurol 64, 414-427.
Clausen T (2003). Na+-K+ pump regulation and skeletal muscle contractility. Physiol Rev 83, 1269-1324.
Fisher WJ & White MJ (1999). Training induced adaptations in the central command and peripheral reflex components of the pressor response to isometric exercise of the human triceps surae. J Physiol 520, 621-628
Hník P, Kríz N, Vyskocil F, Smiesko V, Mejsnar J, Ujec E & Holas M (1973). Work-induced potassium changes in muscle venous effluent blood measured by ion-specific electrodes. Pflugers Arch 338, 177-81.
Jones DA (1996). High and low frequency fatigue revisited. Acta Physiol Scand 156, 265-270.
Jones DA, Bigland-Ritchie B & Edwards RHT (1979). Excitation frequency and muscle fatigue: mechanical responses during voluntary and stimulated contractions. Exp Neurol 64, 414-427.
Lewis T, Pickering GW & Rothschild P (1931). Observations on muscular pain in intermittent claudication. Heart 15, 359-83.
Paterson DJ, Friedland JS, Bacom DA, Clement ID, Cunninham DA, Painter R & Robbins PA (1990). Changes in arterial K+ and ventilation during exercise in normal subjects and subjects with McArdle’s syndrome. J Physiol 429, 339-348.
Ulbricht W (2005). Sodium channel inactivation: Molecular determinants and modulation. Physiol Rev 85, 1271–1301.