
Physiology News Magazine

Glycolysis has many ways to regulate cardiac function
Features
Glycolysis has many ways to regulate cardiac function
Features
Lothar A Blatter (1), Jens Kockskämper (2), Aleksey V Zima (1)
1: Department of Physiology, Loyola University Chicago, Maywood, IL, USA
2: present address: Abteilung Kardiologie und Pneumologie, Georg-August-Universität, Göttingen, Germany
https://doi.org/10.36866/pn.61.36

During every single heart beat each individual cardiac muscle cell consumes substantial amounts of energy in form of ATP. ATP is used for calcium cycling, control of membrane potential and ultimately force development and contraction. At physiological oxygen levels cardiac cells derive 90% or more of the their energy needs from ATP production by mitochondria through oxidative phosphorylation, i.e. less than one tenth of the overall ATP production stems from the glycolytic conversion of glucose to pyruvate and lactate. Nonetheless, glycolysis has been shown to be critical for normal cardiac excitation-contraction coupling (ECC; the sequence of events that links the action potential to Ca release from the sarcoplasmic reticulum (SR) and to muscle contraction). Inhibition of aerobic glycolysis can cause depression of cardiac excitability and action potential-dependent Ca transients, changes in diastolic [Ca]i, and can lead to Ca alternans in cardiac tissue (Fig. 1) (Kockskamper et al. 2005).

The last of these is of particular interest from a pathophysiological point of view because Ca alternans has been identified as a potentially arrhythmogenic abnormality of cardiac Ca signaling (Euler, 1999). Ca alternans is characterized by beat-to-beat alternating large and small amplitude Ca transients, concomitant with electromechanical alternans (i.e. beatto-beat alternations of action potential duration and contraction amplitude). These (sub)cellular phenomena have their clinical equivalents in the form of changes of the electrocardiogram (manifesting e.g. as T-wave alternans or long QT syndrome) or pulsus alternans. These clinically well-documented alterations of the ECG are due to a dispersion of repolarization between different regions of the heart which can lead to unidirectional conduction blocks that can result in re-entrant arrhythmias with potentially lethal consequences for the patient. Despite its pathophysiological and clinical significance, the cellular mechanisms underlying various forms of cardiac alternans are in many aspects still only marginally understood. Evidence, however, is growing that the key to the understanding of cardiac alternans lies in the regulation of cellular energy metabolism and SR Ca release. Recent studies have provided evidence that metabolic inhibition (Huser et al. 2000; Kockskamper & Blatter, 2002) generates conditions that significantly enhance the propensity for Ca and electromechanical alternans. In this context, glycolysis appears to play a central role. As demonstrated recently for atrial tissue, inhibition of glycolysis caused profound Ca alternans with complex subcellular spatio-temporal features, and increased the occurrence of arrhythmogenic Ca waves (Kockskamper & Blatter, 2002).
While a functional link between cellular energy metabolism and SR Ca release during ECC seems obvious, it has remained an intriguing question how glycolysis in particular, while providing quantitatively only a small contribution to the overall cellular energy metabolism, can have such profound effects on cardiac performance. For quite some time now this apparent discrepancy has led to the suggestion that glycolytically-derived ATP may serve as a preferential fuel to maintain, or at least modulate, ion transport pathways. Key glycolytic enzymes have been shown to associate with sarcolemmal and sarcoplasmic reticular membranes and functionally couple to ion transport pathways such as the sarcolemmal KATP channel, voltage-gated Ca channels, the Na/K pump, the Na/H exchange mechanism and the SR Ca pump (SERCA; for references see Kockskamper et al. 2005). The structural and functional prerequisite that would allow the preferential use of glycolyticallyderived ATP over ATP generated by oxidative phosphorylation could reside in a functional microcompartment in which ATP-dependent transporters and channels are tightly regulated by the activity of co-localized glycolytic enzymes, kinases and phosphatases (Goldhaber, 1997). The organization of these enzymes and target proteins into functional complexes (or ‘metabolons’) would allow for self-contained microcompartments in which proteins such as the SR Ca release channel are regulated by locally generated ATP, either directly or through phosphorylation reactions. Indeed, the notion of metabolic microcompartments is not unique to muscle and has also been demonstrated in neurons (for discussion see Huser et al. 2000). Thus the formation of discrete cytosolic microcompartments controlled by localized glycolytic ATP formation may represent a general scheme for the regulation of a number of cellular processes.
Recent experimental evidence has added an additional layer of complexity in the regulation and modulation of SR Ca release by glycolysis. A number of studies (see Kockskamper et al. 2005) have demonstrated that glycolysis affects SR function and SR Ca release not only through generation of ATP but also through direct interactions of glycolytic intermediates and products with the Ca release channel (also known as ryanodine receptor, RyR) itself.
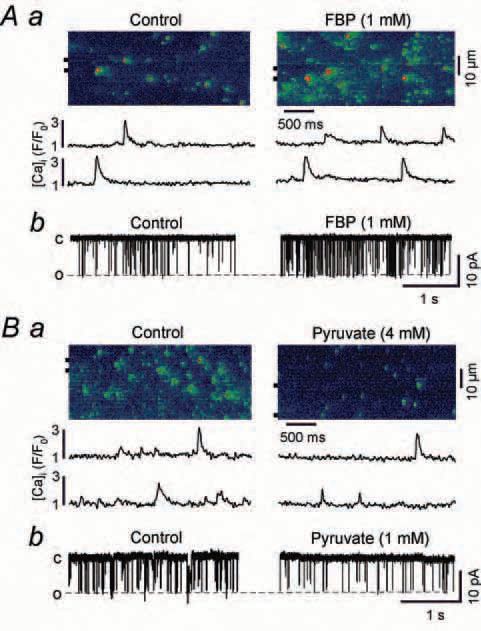
For example (Fig. 2), recording of single channel activity of RyRs incorporated into lipid bilayers revealed that the intermediate of glycolysis fructose-1,6-bisphosphate (FBP) significantly increased, whereas the glycolytic product pyruvate decreased, the open probability of the RyR. Consistent with these findings, spontaneous SR elementary Ca release events (Ca sparks) recorded from atrial myocytes were facilitated by FBP and inhibited by pyruvate.
The current state of our knowledge clearly indicates that modulation of ECC by glycolysis is highly complex. The RyR, the key player responsible for Ca release, is regulated by a number of modulators (ATP, NAD+/NADH, pH, glycolytic intermediates and products) which are directly influenced by glycolytic activity. Thus, any perturbation eliciting only subtle changes of glycolytic flux may, in principle, affect SR Ca release and ECC. At any given time the balance between local changes of stimulatory and inhibitory modulators of the RyR will determine the changes of its open probability.
This becomes particularly evident in acute and chronic cardiac disease states. During acute ischemia, for example, the concentrations of inhibitors of RyR activity rise dramatically (L-lactate, H+, NADH, Mg) whereas, at the same time, the concentrations of stimulators of RyR activity decrease (ATP, NAD+). The overall result will be a decreased activity of the RyR contributing to the suppression of Ca transients during ischemia. In heart failure, transporters and enzymes involved in glycolysis are downregulated (reviewed in Ventura-Clapier et al. 2004) suggesting that altered glycolytic flux may also contribute to abnormal Ca handling and release in the failing heart.
Acknowledgement
This research was supported by NIH R01 HL62331.
References
Euler DE (1999). Cardiac alternans: mechanisms and pathophysiological significance. Cardiovasc Res 42, 583-590.
Goldhaber JI (1997). Metabolism in normal and ischemic myocardium. In The Myocardium, 2nd edn, ed. Langer GA, pp 325393. Academic Press, San Diego.
Huser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL & Blatter LA (2000). Functional coupling between glycolysis and excitationcontraction coupling underlies alternans in cat heart cells. J Physiol 524, 795-806.
Kockskamper J & Blatter LA (2002). Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J Physiol 545, 65-79.
Kockskamper J, Zima AV & Blatter LA (2005). Modulation of sarcoplasmic reticulum Ca2+ release by glycolysis in cat atrial myocytes. J Physiol 564, 697-714.
Ventura-Clapier R, Garnier A & Veksler V (2004). Energy metabolism in heart failure. J Physiol 555, 1-13.