
Physiology News Magazine

Hepcidin, body iron and infection
Features
Hepcidin, body iron and infection
Features
Ted Debnam & Kaila Srai
Departments of Physiology and Biochemistry & Molecular Biology, University College London, UK
https://doi.org/10.36866/pn.61.38


Iron deficiency anaemia is the most common nutritional disorder affecting over 30% of the world’s population. In developing countries the condition is frequently exacerbated by infection. There is no excretory pathway for iron and body iron status therefore depends on the efficacy of dietary iron uptake. This process is suppressed by hepcidin, a liver-derived peptide that is also an antimicrobial agent.
Adequate levels of iron are central to tissue function but the metal is toxic in excess. A healthy 70 kg adult contains 3-4 g iron, most being a constituent of haemoglobin or myoglobin. Each day round 20 mg of body iron is recycled through reticuloendothelial macrophages in spleen, liver and bone marrow, this being a consequence of normal erythrocyte turnover.
By comparison, the daily obligatory body loss of iron (via urine, GI tract and skin) is small, some 1-2 mg. Nonetheless, if this is not replaced by duodenal uptake of an equivalent amount of dietary iron, the inevitable long term consequence will be iron deficiency. Only about 10% of dietary iron needs to be absorbed by the small intestine to match obligatory losses, but the body has the capacity to increase this proportion in times of higher demand, e.g. pregnancy, or to meet the requirements for enhanced erythropoiesis during haemorrhage or hypoxia. Conversely, the response to increased body iron is reduced intestinal iron uptake. Although key elements in the mechanism for iron absorption across duodenal absorptive cells (enterocytes) are known, the way in which the overall process adapts to altered physiological demands is less clear.
The first link between inflammation and intestinal iron transport came from studies using the turpentine abscess technique to induce an inflammatory state. The procedure reduced duodenal iron transport in mice (Raja et al. 1990). A major thrust came with the identification of hepcidin in urine and plasma during a search for novel antimicrobial peptides (Ganz, 2003). Hepcidin, also called liver-expressed antimicrobial peptide 1 (LEAP-1), is synthesized by hepatocytes and is now known to be crucial for body iron homeostasis by acting as a negative regulator of iron exit from enterocytes and reticuloendothelial macrophages (Fig. 1). An injection of turpentine increases the expression of hepcidin by 6-fold, and lowers circulating iron level in wild type mice but not in animals unable to express hepcidin. The strength of the evidence linking this 25 amino-acid peptide to iron transport is now so great that it has acquired endocrine status. There is a reciprocal relationship between the rate of intestinal iron transport and expression of hepcidin. Thus conditions such as dietary iron deficiency, hypoxia and haemorrhage are associated with decreased hepcidin expression and this leads to adaptive increases in iron absorption. Conversely, hepcidin expression is increased by iron overload and less dietary iron crosses the duodenum. We recently showed that injection of synthetic hepcidin reduces duodenal iron absorption in normal and iron deficient mice (Laftah et al. 2004). Others observed that deletion of the hepcidin gene promotes iron transport. The finding that hepcidin reduces iron transfer across cultured intestinal epithelium provides compelling evidence for a direct action of the peptide on this tissue.
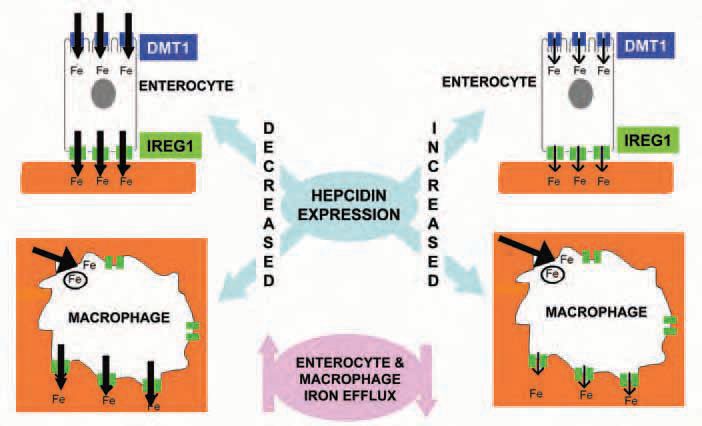
How does hepcidin achieve its action on enterocytes? There is growing evidence that the peptide promotes the internalization and degradation of Ferroportin (IREG1), the protein necessary for iron transfer to the blood (Fig. 1). At the brush border, hepcidin also reduces expression of divalent metal transporter 1 (DMT1), which mediates iron uptake from the gut lumen. However, it is unclear whether this response represents a direct action of hepcidin or is a consequence of changes in enterocyte iron level resulting from reduced Ferroportin-mediated iron efflux. The mucosal hepcidin receptor has yet to be characterized but some suggest that it may be Ferroportin itself.
The functional link between changes in body iron, hepcidin and duodenal iron uptake is unclear. Recent work suggests that altered hepcidin synthesis in response to an increased or decreased level of body iron involve the hepatic sensing of plasma transferrin iron saturation by a process involving TfR2 in concert with other proteins including TfR1, haemojuvelin and HFE (Frazer & Anderson, 2003). Our observation that hypoxia reduces hepcidin expression both in vivo and in isolated hepatocytes (Leung et al. 2005) implies that hepcidin secretion is also linked to hepatic sensing of oxygen (Fig. 2). Inflammation is another recognized stimulus of hepcidin expression, an effect mediated by IL-6, but not TNF-α (Krause et al. 2000) indicating that hepcidin is a type II acute phase reactant.

The importance of iron and oxygen sensing linked to hepcidin expression and altered enterocyte iron transport is easy to appreciate. However, what is the survival advantage conferred by raised levels of hepcidin during inflammation? Interestingly, the peptide also reduces iron exit from macrophages by an action on Ferroportin, a process akin to that in enterocytes (Fig. 1). Macrophages can be regarded as reservoirs for iron, releasing the metal in times of increased body demand whilst storing iron to prevent circulating levels reaching toxic levels. The significance of the complementary effects of hepcidin on macrophages and enterocytes may be related to the requirement of iron for bacterial growth. By reducing iron efflux from macrophages and enterocytes, hepcidin will restrict iron availability to proliferating bacteria. Anaemia seems to be the price to pay for the fight against infection.
The anaemia of chronic renal failure (CRF) is usually attributed to inappropriate erythropoietin (Epo) secretion from a reduced kidney mass, but information suggests that hepcidin may also be involved. CRF patients receiving Epo often require iron supplementation to satisfy erythropoietic demands. However, oral iron tends to be less effective than systemic iron administration implying that intestinal transport of iron is inadequate. Interestingly, inflammation frequently accompanies CRF, particularly in patients on dialysis; not surprisingly, these patients tend to have raised hepcidin secretion. The peptide may therefore override Epo action in renal failure by acting on macrophages and enterocytes to reduce the availability of iron to developing red blood cells, culminating in iron deficiency anaemia.
A greater understanding of ways by which the liver influences iron handling by enterocytes and iron storage cells will throw light on the role of hepcidin in iron homeostasis in health and disease. Such knowledge may ultimately lead to tissue-specific therapies for more effective management of anaemia and other pathological changes in body iron.
Acknowledgements
Iron research in the authors’ laboratories is currently funded by the BBSRC, the Roche Foundation for Anaemia Research and the National Kidney Research Fund.
References
Frazer DM & Anderson GJ (2003). The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Mol Dis 30, 288-297.
Ganz T (2003). Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 102, 783-788.
Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K (2000). LEAP-1, a novel highly disulfidebonded human peptide, exhibits antimicrobial activity. FEBS Lett 480,147-150.
Laftah AH, Ramesh B, Simpson RJ, Solanky N, Bahram S, Schumann K, Debnam ES, Srai SKS (2004). Effect of hepcidin on intestinal iron absorption in mice. Blood 103, 3940-3944.
Leung PS, Srai SK, Mascarenhas M, Churchill LJ, Debnam ES (2005). Increased duodenal iron uptake and transfer in a rat model of chronic hypoxia is accompanied by reduced hepcidin expression. Gut 54, 1391-1395.
Raja KB, Duane P & Peters TJ (1990). Effects of turpentine-induced inflammation on the hypoxic stimulation of intestinal Fe3+ absorption in mice. Int J Exp Pathol 71, 785-789.