
Physiology News Magazine

HYPOGLYCAEMIA AND CENTRAL WHITE MATTER: MECHANISMS OF INJURY AND NEUROPROTECTIVE STRATEGIES
The brain is vulnerable to low blood glucose, and hypoglycaemia is a major problem for diabetics. Here Angus Brown explains how this hypoglycaemia may cause cellular damage to the white matter.
Features
HYPOGLYCAEMIA AND CENTRAL WHITE MATTER: MECHANISMS OF INJURY AND NEUROPROTECTIVE STRATEGIES
The brain is vulnerable to low blood glucose, and hypoglycaemia is a major problem for diabetics. Here Angus Brown explains how this hypoglycaemia may cause cellular damage to the white matter.
Features
Angus Brown
Department of Neurology, University of Washington, Seattle
https://doi.org/10.36866/pn.43.17
Although the brain comprises only 2% of body weight, it accounts for 75% of the body’s daily requirement for glucose. This high metabolic demand requires an uninterrupted blood supply, which renders the brain vulnerable to shortfalls in delivery of blood-borne glucose. This critical dependence of the human central nervous system (CNS) on glucose has led to the evolution of complex homeostatic mechanisms that maintain blood glucose concentrations within strictly regulated euglycaemic (normal) levels, mainly by the actions of insulin and glucagon. However, this homeostatic system can be disrupted by disease, leading to life-long problems of “managing” blood glucose levels.
Type-1 diabetes, hyper and hypoglycaemia
People suffering from Type-I (insulindependent) diabetes are unable to manufacture insulin due to autoimmune destruction of pancreatic Beta-cells. Since insulin maintains blood glucose in the euglycaemic range by facilitating transport of glucose from blood into cells, patients with Type-I diabetes cannot maintain blood glucose at euglycaemic levels, and periods of both hyperglycaemia (higher than normal blood glucose) and hypoglycaemia (lower than normal blood glucose) are inevitable. Hyperglycaemia in particular is implicated in the long-term morbidity (neuropathy, retinopathy and nephropathy) that typically afflicts Type-I diabetics, and the main goal of diabetic therapy is thus to prevent hyperglycaemia by introduction – typically by injection- of exogenous insulin. However, it is a cruel fact that diabetics who actively strive to maintain euglycaemia often fall victim to hypoglycaemia, due to the difficulties of matching insulin delivery to minute-by-minute blood glucose concentration. Indeed, fear of hypoglycaemic episodes is the major deterrent that prevents diabetics from maintaining a rigid regime of insulin therapy and achieving euglycaemia.

While the avoidance, and complications, of hyperglycaemia have received the majority of attention, it is increasingly appreciated that hypoglycaemia is a major factor in decreased quality of life for most diabetics and their families. This reflects the need for continual hypoglycemia avoidance strategies, as we] I as the potentially serious consequences of suffering a severe hypoglycaemic episode. When blood glucose falls to hypoglycaemic levels (defined by the British Diabetic Association as below 4 mmol/1) there is a series of responses, associated with both autonomic and neurological symptoms, whose intensity increases with the degree of hypoglycaemia (Figure 1). The detection and correct interpretation of responses to mild hypoglycaemia can act to warn the patient of an impending hypoglycaemic episode, and therefore to take suitable counter-measures. However, hypoglycaemia is not always detectable, for a variety of reasons ( e.g. hypoglycaemia unawareness, nocturnal onset), and all Type-1 diabetics will inevitably endure hypoglycaemic episodes. It is estimated that the typical Type-I diabetic will endure 70 episodes severe enough to require assistance, and 3500 mild episodes, of hypoglycaemia in his/her lifetime [l]. There is increasing evidence that the cumulative effects of these repeated hypoglycaemic episodes eventually result in permanent CNS injury, reflected in, among other deficits, impaired memory and decreased cognitive ability.
Mechanism of white matter injury
It is commonly accepted that the brain contains no endogenous energy supplies, and relies solely on the blood for nutrients. However, a recent study from our laboratory, using the adult rat optic nerve (RON) as a model of central white matter, demonstrated that central white matter does contain a utilizable energy source in the form of glycogen within astrocytes [2]. The astrocytic glycogen is metabolized to .lactate, which is then transported into axons where it sustains function for over 30 minutes after withdrawal of glucose (Figure 2). Beyond this point glycogen stores are exhausted, and axon function fails. We were able to pharmacologically up-and down-regulate glycogen levels resulting in, respectively, increased and decreased latency to axon failure during hypoglycaemia. These results are encouraging, since they suggest that at least certain parts of the CNS are not solely at the mercy of blood to deliver nutrients, but can use an endogenous energy supply to maintain function during hypoglycaemic episodes. This important role for glycogen in preventing hypoglycemiainduced CNS injury is being pursued in our laboratory.
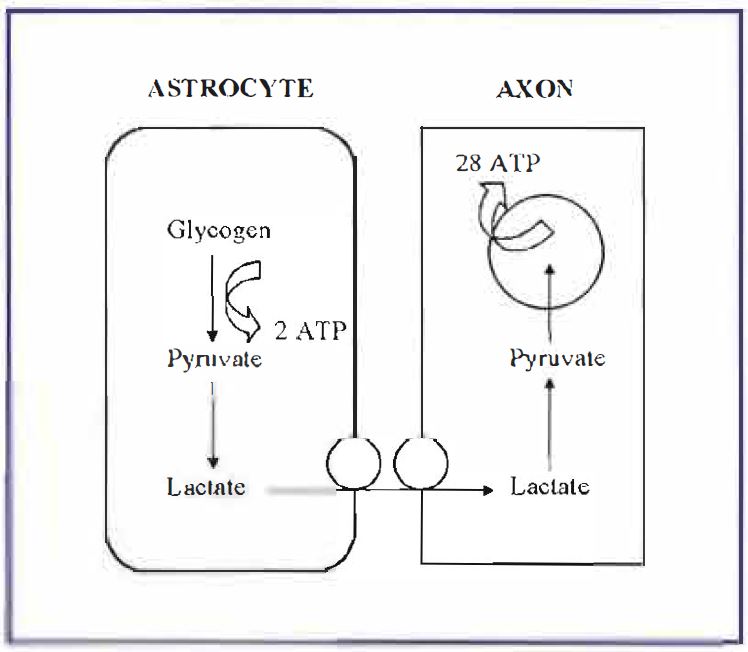
Once astrocytic glycogen energy reserves are exhausted during a hypoglycaemic episode, CNS injury is inevitable. Almost all of the previous studies on the effects of hypoglycaemia on the CNS have concentrated on gray matter areas, where glutamate excititoxicity appears to be the major cause of neuronal death [3]. In contrast, almost nothing 1s known about how hypoglycaemia affects white matter areas, even though they comprise 50% of brain volume and are fundamental to brain integrity. We found that a 1-hour period of hypoglycaemia (i.e. no glucose in the artificial CSF bathing the RON), followed by a I hour recovery period in control artificial CSF containing 10 mM glucose, resulted in approximately 50% reduction in RON function, indicating that irreversible injury had occurred. Neuronal function was assessed by recording the stimulus-evoked compound action potential (CAP) from the RON. Repeating the experiment in a Ca2+-free artificial CSF resulted in no functional loss, implying that hypoglycaemiainduced injury is a Ca2+ -dependent process [4]. The lack of glutamatergic synapses in white matter implies a different mechanism of cell death from the glutamate excitotoxicity in gray matter. In order to evaluate Ca2+ movements during hypoglycaemia, we recorded extracellular free Ca2+ using Ca2+ -sensitive microelectrodes. Extracellular [Ca2+] fell with a similar time course to the progressive failure of the CAP, suggesting that Ca2+ moves into intracellular compartments as cellular injury occurs. Immunocytochemical studies demonstrated that the RON contains exclusively L-type Ca2+ channels, which are located on both axons and astrocytes [5]. Application of L-type Ca2+ channel blockers during the hypoglycaemic insult caused decreased Ca2+ influx and was neuroprotective, implicating L-type Ca2+ channels as a route of toxic Ca2+ influx. Inhibitors of Na+-Ca2+ exchange similarly decreased Ca2+ influx and were neuroprotective.
It thus appears that toxic Ca2+ influx, presumably directly into axons via L-type Ca2+ channels and reverse operation of the Na +-Ca2+ exchanger (Figure 3) mediates hypoglycaemic axon death in this preparation [4].

Future directions
The ultimate goal for research into Type-I diabetes is the correction of insulin deficiency. Eventually this is likely to be achieved by introduction of viable insulin-secreting cells into patients, whether by implanting clusters of insulin-secreting cells, by stem cell technology or by gene therapy. However, the successful clinical introduction of these technologies is at least several years in the future. Until a cure for Type 1 diabetes is a reality, research must continue into areas that will improve the quality of life for diabetic patients. These include improving glucose sensing technology, and developing short-acting insulin, inhaled insulin and insulin analogues to help maintain euglycaemia. In addition, investigating the mechanism(s) by which hypoglycaemia injures the CNS at the cellular level is a priority. This will hopefully allow development of neuroprotective strategies that will spare CNS function during the inevitable periods of hypoglycaemia endured by all Type-1 diabetics.
References
1. Frier, B.M., Fisher, B.M. (1999). Hypoglycaemia in Clinical Diabetes.
2. Wender, R., Brown, A.M., Fern, R., Swanson, R.A., Ransom, B.R. (2000). J Neurosci 20, 6804-6810.
3. Monyer, H .. Goldberg, M.P., Choi, D.W. (1989). Brain Res 483, 347-354.
4. Brown, A.M., Wender, R., Ransom, B.R. (2001). J Cereb Blood Flow Metab. 21, 385-395.
5. Brown, A.M., Westenbroek, R.E., Catterall, W.A., Ransom, B.R. (2001). J Neurophysiol 85, 900-911.