
Physiology News Magazine
Imaging Molecular Dynamics for Drug Discovery
In the fight against cancer, Kurt Anderson and his collaborators are using advanced imaging to characterise drug response
Features
Imaging Molecular Dynamics for Drug Discovery
In the fight against cancer, Kurt Anderson and his collaborators are using advanced imaging to characterise drug response
Features
Max Nobis, David Herrmann & Paul Timpson
Garvan Institute for Medical Research, Sydney, Australia
Zahra Erami
Albert Einstein College of Medicine, New York, USA
Ewan McGhee
Beatson Institute for Cancer Research, Glasgow, UK
Kurt Anderson
The Francis Crick Institute, London, UK
https://doi.org/10.36866/pn.104.27
Since the discovery of the green fluorescent protein (GFP) in the early 1990s, the properties of fluorescent proteins have been greatly improved through genetic manipulation of the tri-peptide chromophore at the heart of the protein. Early improvements led to probes which were more monomeric, brighter, more photo-stable, and had a broader spectral range (reviewed in Muller-Taubenberger & Anderson, 2007).
Fluorescent proteins have been used in a variety of approaches to develop biosensors, which use fluorescence to read out the activation status of cellular signalling pathways. In the simplest case, knocking an FP into an endogenous gene locus can be used as a reporter of gene expression. FPs can also be coupled to transcription factors such as NF-kB, which translocates in and out of the nucleus in response to upstream signals such as TNFa (Nelson et al., 2004). Photo-bleaching of FPs can be used to report and quantify the dynamics of structural proteins such as E-cadherin (Erami et al., 2015). Finally, FPs can be used to detect conformational changes within proteins on the basis of Fluorescence Resonance Energy Transfer (FRET).
FRET is the non-radiative transfer of energy between two fluorophores having overlapping emission and excitation spectra, the proper orientation, and proximity of approximately 3 – 5 nm. Such probes can reveal unexpected feedback mechanisms by which cancer cells evade therapeutic treatment (Hirata et al., 2015). FRET can be most accurately detected as a shortening of the fluorescence lifetime of the donor fluorophore. Recently, even this property has been targeted for optimisation, resulting in FPs having very long fluorescence lifetimes in order to expand the dynamic range of
FRET-based biosensors (Goedhart et al., 2012). Alternatively, dark acceptor FPs have been engineered which are able to accept energy from a FRET donor without themselves emitting light (Ganesan et al., 2006; Pettikiriarachchi et al., 2012),. Such fluorophores facilitate the multiplexing of FRET probes to read out the states of multiple signal transduction pathways simultaneously, which is increasingly important for understanding the responses of complex biological systems (Machacek et al., 2009).
The cost of developing a new drug doubles approximately every 9 years (Nosengo, 2016). This crisis in drug discovery requires innovative new approaches at all stages of the discovery pipeline, but especially at the later stages when failure becomes expensive. Modern high-throughput screening approaches are effective at identifying compounds, which elicit a specific cellular response in cells grown on glass coverslips. Such screens typically rely on single read-out biomarkers, although newer approaches may rely on multi-parametric phenotypic readouts such as morphology, migration, and cell cycle progression (reviewed in Conway et al., 2014).
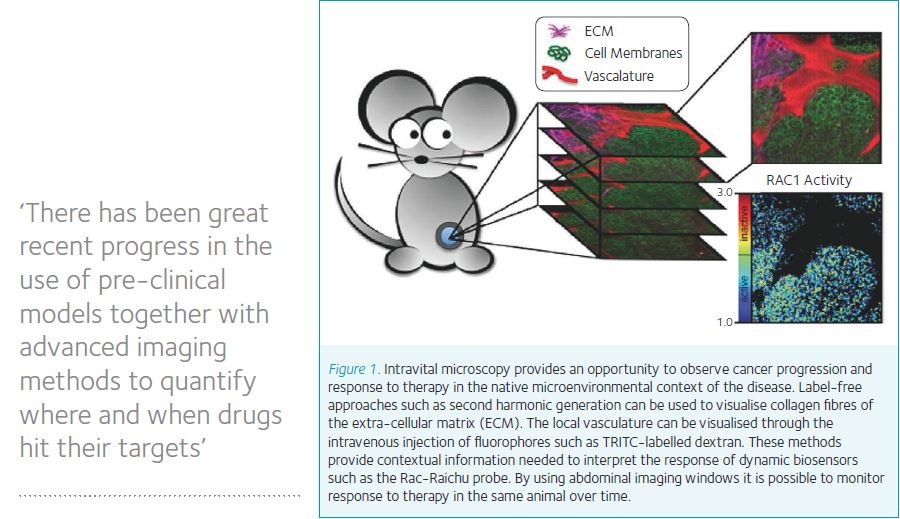
Unfortunately, there is a high attrition rate for progression of drug candidates from cell-based assays into the later stages of pre-clinical validation and clinical studies. We therefore urgently need better disease models on which to base predictions of clinical efficacy, and better tools with which to interrogate therapeutic response within these pre-clinical models. FRET-based biosensors have proven to be useful for cell-based assays, and more recently have been introduced into pre-clinical cancer models to facilitate the investigation of the spatial and temporal dynamics of drug action in unprecedented detail. This approach has benefitted from improvements in the design and use of surgically implanted imaging windows, which facilitate longitudinal studies in which the same animal is imaged over time. This approach improves the quality of data by reducing biological noise and thereby reduces the number of animals required to obtain a time course (eg of drug response), both of which are important aspects of the 3Rs (Burden et al., 2015). Previous imaging windows were commonly restricted to use on the skin flap of nude mice (Lehr et al., 1993), but recently chambers have been designed which are well tolerated over long periods of time and enable the imaging of subcutaneous organs such as breast and intra-peritoneal organs such as liver, intestine, and pancreas (Ritsma et al., 2013).
There has been great recent progress in the use of pre-clinical models together with advanced imaging methods to quantify where and when drugs hit their targets. This work depends on the use of a pipeline of complementary model systems, including cultured cells, 3D culture models, organoids, ex vivo tissue, transplantation models, and full genetic models of disease (reviewed in (Timpson et al., 2011).
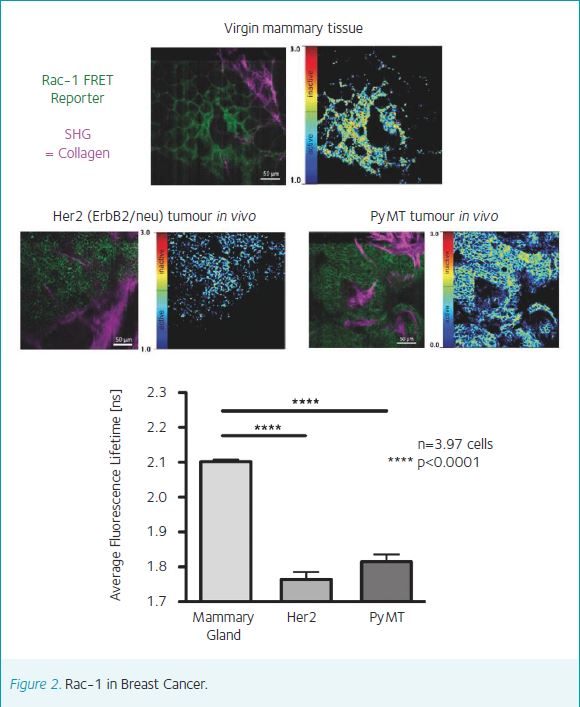
In a pioneering study we used FRAP to show that the dynamics of E-cadherin in cell-cell junctions were different for the same cancer cells grown in vitro on glass coverslips and in vivo as subcutaneous tumors (Serrels et al., 2009). Crucially, the response of E-cadherin to drug treatment was completely different in the two different cellular environments. In another approach, we have shown that FLIM-FRET is sufficiently sensitive to detect sub-cellular responses to drug treatment in vivo. Treatment of mice bearing subcutaneous PDAC tumours, with a dose of Dasatinib sufficient to inhibit PDAC metastasis, specifically inhibited Rho GTPase in the tips of cells rather than globally throughout the cell body (Timpson et al., 2011). Additional work has shown that intra-vital FLIM-FRET can be used to characterise drug response in different regions of subcut PDAC tumours, and with respect to the tumour vasculature (Nobis et al., 2013).
Now, our work using FRAP of E-cadherin has come full-circle with the production of a mouse expressing GFP-E-cadherin from the Hprt locus. This mouse enables the visualization of E-cadherin dynamics in a wide variety of tissues including liver, breast, kidney and pancreas, and has been crossed into the KPC model of pancreatic cancer to demonstrate the specific mobilization of E-cadherin in primary PDAC tumours in response to mutant p53 (Erami et al., 2016). Thus, there is an emerging body of work, which clearly demonstrates the importance for drug discovery of imaging molecular dynamics in pre-clinical models.
References
Muller-Taubenberger A. & Anderson KI (2007). Recent advances using green and red fluorescent protein variants. Appl Microbiol Biotechnol 77, 1-12
Nelson DE et al., (2004). Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306, 704-708
Erami Z, Timpson P, Yao W, Zaidel-Bar R & Anderson KI (2015). There are four dynamically and functionally distinct populations of E-cadherin in cell junctions. Biol Open 4, 1481-1489
Hirata E et al., (2015). Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 27, 574-588
Goedhart J et al., (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun 3, 751
Ganesan S, Ameer-Beg SM, Ng TT, Vojnovic B & Wouters FSA (2006). Dark yellow fluorescent protein (YFP)-based Resonance Energy-Accepting Chromoprotein (REACh) for Forster resonance energy transfer with GFP. Proc Natl Acad Sci U S A 103, 4089-4094
Pettikiriarachchi A, Gong L, Perugini MA, Devenish RJ & Prescott M (2012).Ultramarine, a chromoprotein acceptor for Forster resonance energy transfer. PLoS One 7, e41028
Machacek M et al., (2009). Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99-103
Nosengo N (2016). Can you teach old drugs new tricks? Nature 534, 314-316
Conway JR, Carragher NO & Timpson P (2014). Developments in preclinical cancer imaging: innovating the discovery of therapeutics. Nat Rev Cancer 14, 314-328
Burden N, Chapman K, Sewell F & Robinson V (2015). Pioneering better science through the 3Rs: an introduction to the national centre for the replacement, refinement, and reduction of animals in research (NC3Rs). J Am Assoc Lab Anim Sci 54, 198-208
Lehr HA, Leunig M, Menger MD, Nolte D & Messmer K (1993). Dorsal skinfold chamber technique for intravital microscopy in nude mice. Am J Pathol 143, 1055-1062
Ritsma L et al., (2013). Surgical implantation of an abdominal imaging window for intravital microscopy. Nat Protoc 8, 583-594
Timpson P, McGhee EJ & Anderson KI (2011) Imaging molecular dynamics in vivo – from cell biology to animal models. J Cell Sci 124, 2877-2890
Serrels A. et al., (2009). Real-time study of E-cadherin and membrane dynamics in living animals: implications for disease modeling and drug development. Cancer research 69, 2714-2719
Timpson P. et al., (2011). Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer research 71, 747-757
Nobis, M. et al., (2013). Intravital FLIM-FRET imaging reveals dasatinib-induced spatial control of src in pancreatic cancer. Cancer research 73, 4674-4686
Erami, Z. et al. (2016). Intravital FRAP Imaging using an E-cadherin-GFP Mouse Reveals Disease- and Drug-Dependent Dynamic Regulation of Cell-Cell Junctions in Live Tissue. Cell Rep 14, 152-167