
Physiology News Magazine

Limits to human performance caused by muscle fatigue
David Allen and colleagues describe the mechanisms of fatigue in muscles and show how they differ in various types of activity
Features
Limits to human performance caused by muscle fatigue
David Allen and colleagues describe the mechanisms of fatigue in muscles and show how they differ in various types of activity
Features
David Allen
Department of Physiology and Institute for Biomedical Research, University of Sydney, Australia
Jan Lännergren & Håkan Westerblad
Department of Physiology and Pharmacology Karolinska Institutet, Stockholm, Sweden
https://doi.org/10.36866/pn.53.7

An elite athlete can run 100 m in 10s; naively one might imagine that the same athlete could run 1000 m in 10 x 10 = 100 s but in fact the world record is around 130 s. Similarly over 10,000 m the world record is not 1000 s but about 1600 s so again we find that over a longer distance performance declines. In this respect the human machine is very different to many other machines we are familiar with; for instance, once a car has accelerated to its maximum speed it can normally maintain that speed indefinitely until its supply of fuel runs out and then it simply stops; there is no period of gradually declining performance as the fuel is consumed as is seen with muscle performance. This difference between muscles and other machines suggests that there may be evolutionary advantages to a period of gradually declining performance; it might signal to the animal that fuel is starting to run out and that, for instance, an alternative strategy for escaping a predator might need to be considered.
In this article we show that the main cause of this decline in performance is located in the muscles and is associated with the consumption of energy sources. While energy sources decline, by-products tend to accumulate and some of these inhibit the contractile proteins and contribute to the reduced force. Moreover, one important cause of muscle fatigue is that calcium release, which normally triggers contraction, starts to fail in fatigued muscles.
Where is the site of fatigue?
Muscle contraction is a complex chain of events (Fig. 1). Voluntary contraction commences in the motor cortex and a volley of action potentials (i) activates the α-motor neuron (ii) which excites the muscle through the neuromuscular junction (iii). The muscle action potential is transmitted along the surface membrane (iv) and down the T-tubules (v). A voltage sensor in the T-tubules causes the opening of Ca2+ release channels in the sarcoplasmic reticulum (SR) (vi) causing a rise in myoplasmic [Ca2+]i . Ca2+ binds to troponin (vii), activating crossbridge cycling and force (viii) and relaxation requires the reuptake of Ca2+ into the SR (ix). To understand the mechanism of fatigue we need to identify the links in this chain which fail during repeated activity. One of the earliest investigations was the classic experiments of Merton (1954). The subject contracts a small muscle of the thumb repeatedly until the force produced is reduced to half, which takes a few minutes. Then the ulnar nerve, which supplies the muscle and lies just under the skin, is stimulated with external electrodes. If fatigue were in the brain or spinal cord, it should be possible to overcome it by stimulating the motor nerve – in practice, however, nerve stimulation usually has little effect on the force produced. To obtain this result the subject must be well motivated and indifferent to the pain of the fatigued muscle; obviously these conditions do not always apply particularly outside laboratory experiments. Thus, in well motivated subjects fatigue is mostly in the muscle itself and is conveniently studied in isolated muscles (*1)

*1 For more discussion on mental fatigue see the article by Mads Dalsgaard on page 29
Why do muscles fatigue?
Muscles may fatigue for many different reasons. The CNS may fail to produce action potentials, the muscle action potential may fail because of ionic changes, energy stores can run out, reactive oxygen species may damage proteins, there may be structural damage to muscles, etc. The traditional explanation for fatigue is that the breakdown of muscle glycogen, a major store of energy inside muscle fibres, leads to the accumulation of lactic acid which impairs muscle function. It is certainly true that in some types of fatigue intracellular lactic acid accumulates and that in experiments on isolated contractile proteins (skinned fibres) acidosis can inhibit the contractile proteins. However, there are other types of fatigue in which lactic acid does not accumulate and a recent finding is that when the effects of acidosis were examined at body temperature rather than at room temperature, the inhibitory effects were minimal. Thus the lactic acid theory of muscle fatigue is losing support, though this is not yet reflected in many text book accounts (Westerblad et al. 2002).
We need first to consider the energy sources inside muscle fibres. ATP is the universal source of energy inside cells and is used directly by the crossbridges and ion pumps. However, there is only enough ATP present to power a maximal contraction for 2–3 s. When a muscle is completely depleted of ATP, it cannot contract and becomes very stiff (rigor); if this were ever to occur in normal activity it would cause severe muscle damage and this may be one of the evolutionary pressures encouraging the development of fatigue. To avoid ATP depletion muscle has a variety of backup sources of energy and these are so effective that the bulk ATP concentration never falls below about 20 % of the resting level even in maximally fatigued muscles. To aid understanding we quote for each source of energy the period of time it could power maximal contraction recognising that this rarely occurs and that generally several pathways will be activated in parallel. An easily accessible backup source is phosphocreatine which directly recharges ATP and this supply lasts for 10–20 s. Another important source of energy is glycogen which is stored in the muscle and therefore readily accessible. Glycogen can be broken down anaerobically and used in this way would last only 2-3 min. The sources of energy so far mentioned do not require oxygen (anaerobic pathways) and are the only sources of energy in very brief maximal activities or when the oxygen supply is not available. Alternatively glycogen can be broken down aerobically. Although this process is markedly slower than the anaerobic breakdown, it produces enough ATP to keep the muscle contracting at a near maximal rate for 30–60 min. ATP can also be produced from the aerobic metabolism of fat stores, which are very large but can only be metabolised relatively slowly. Once the glycogen stores are depleted, muscles must rely on fat metabolism.
The 100 m sprint
The running speed during a 100 m sprint is much higher than during longer runs; the short duration means that fatigue is less of a problem. However, even during short sprints there is some fatigue and the maximum running speed in a 100 m sprint occurs after about 60 m. Why does the running speed decline during such a short sprint? The answer to this is not entirely clear. It is, however, clear that lactic acid has little to do with it. Relatively little lactic acid is formed during such a short activity. Instead most of the energy comes from breakdown of phosphocreatine. Breakdown of phosphocreatine consumes hydrogen ions so the net effect is that myoplasmic pH is not significantly altered during the sprint. One product of phosphocreatine breakdown is phosphate ions and these have been shown to depress muscle function: they reduce both Ca2+-sensitivity of the contractile proteins (Fig. 1 vii) and the ability of the contractile proteins to produce force (Fig. 1 viii). Thus phosphate ion accumulation is probably an important contributor to fatigue during a 100 m sprint.
While phosphocreatine breakdown may contribute to fatigue by producing phosphate ions, it is also the fastest provider of ATP to crossbridges and ion pumps which makes it possible for these to work at a high rate. Without a rapid ATP supply, ADP will accumulate and this will slow down crossbridge cycling and ion pumping and hence decrease the power output of the muscle. In recent years it has become increasingly popular for sprinters to ingest enormous amounts of creatine and this seems to have a small beneficial effect on the performance in sprint running – but for elite athletes even a very small improvement can make the difference between winning and losing. Excessive creatine intake results in increased levels of phosphocreatine in muscles so that the period of close to maximal performance is prolonged.
Continuous maximal contraction
A continuous maximal contraction is needed when lifting something very heavy, like a piano. Everyone will be aware of how rapidly fatigue can set in during such activities. In this situation the muscle machinery is going at full speed and energy is consumed at a rapid rate. In addition, the blood flow to the active muscle(s) is stopped during maximal contractions so that no delivery of oxygen (to support muscle contraction) or removal of metabolites or ions will occur. Thus severe fatigue develops within seconds and the muscle becomes rapidly weaker. Changes of the ionic distribution over the cell membrane probably contribute to this type of fatigue. Each action potential is associated with entry of sodium ions into the cell and exit of potassium ions from the cell; consequently potassium ions tend to accumulate outside of the fibres and this results in depolarization and impaired electrical activation of muscle cells. This extracellular accumulation of potassium is likely to be larger in the narrow lumen of the t-tubules (see Fig. 1) from which the potassium ions can only diffuse rather slowly. This leads to impaired propagation of action potentials into the deep parts of fibres and, as a consequence, reduced Ca2+ levels and contractile parts of fibres and, as a consequence, reduced Ca2+ levels and contractile activation in the central core of fibres (Westerblad et al. 1990).
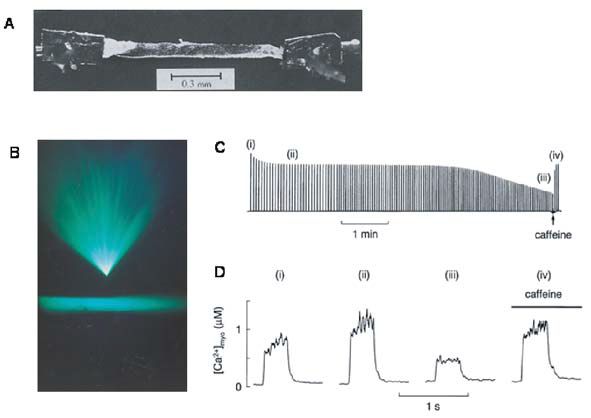
The 5 km race
Events of this sort last 10 or so minutes and are performed at quite close to the maximum capacity of the muscles and involve both the aerobic and the anaerobic ATP pathways. In isolated muscle fibres stimulated until fatigue there is a prominent failure of Ca2+ release from the SR (see Fig. 2D). It is easy to show that this decline of Ca2+ release is important in the failing muscle performance because caffeine can increase SR Ca2+ release and overcome (temporarily) much of the decline of force (Fig. 2C & D). Fortunately, this effect of caffeine occurs at concentrations about 1000 times greater than achieved in muscles after normal ingestion of tea or coffee. The exact cause of the failure of Ca2+ release is not known but it probably involves a metabolic component because it is accelerated by inhibiting oxidative phosphorylation. A recent hypothesis is that SR Ca2+ release may fail because Ca2+ and phosphate inside the SR exceed the solubility product of Ca2+ phosphate and precipitate, thereby reducing the free Ca2+ available for release. This mechanism only operates because myoplasmic phosphate rises substantially during fatigue and is capable of entering the SR (for review see Allen & Westerblad, 2001).
Running longer distances
Anyone who has tried running a marathon will be aware of the overwhelming muscle weakness experienced in the final stages of the race. This type of fatigue develops quite suddenly, known as ‘hitting the wall’, and correlates with the near final depletion of glycogen in muscles. However, the precise reason why glycogen-depleted muscles feel weak remains something of a mystery. Muscle biopsies show that at the end of a marathon glycogen is depleted but ATP is only marginally reduced and studies on isolated muscle proteins show that this small reduction of ATP does not affect contractile performance. Lactic acid accumulation is minimal under these circumstances, because lactic acid can leave the cell after a few minutes, and phosphate accumulation is also only moderate. Instead the main factor seems to be failure of calcium release inside the cells which is associated with the glycogen depletion. New insights into this have recently come from skinned fibre experiments which also show a failure of Ca2+ release around the time the glycogen is depleted (Stephenson et al. 1999). However, in these experiments both ATP and PCr were present in the solutions applied to the skinned muscle and should prevent any metabolic consequences of glycogen depletion. Instead it has been suggested that glycogen may exert a structural role, especially since some of the most labile glycogen in muscle is located at T-tubular/SR junction. Because glycogen is so central to muscle performance, many diets and training procotols have been devised which increase muscle glycogen and such diets can cause a pronounced improvement in performance for activities lasting 1–2 hours.
Recovery from fatigue
Recovery from fatigue has been found to be complex, with both fast and slower components. The faster component is due to reversal of the metabolic changes which caused fatigue in the first place; for example, wash-out of lactic acid and restoration of the phosphocreatine store which will take up the excess of phosphate ions. These processes are relatively fast and are completed in minutes. There remains a second component of fatigue which recovers much more slowly, taking several days for muscles to regain their normal capacity. In real life we will experience this as ‘heavy’ legs: we can perform almost normally but this requires markedly more mental effort (i.e. the CNS has to produce action potentials at a higher frequency to obtain the same force).
Experiments on isolated muscles suggest that this delayed recovery is also caused by reduced Ca2+ release (Westerblad et al. 2000). The action potential is normal and the SR is normally loaded with Ca2+ but the coupling between the action potential and Ca2+ release is damaged. One suggestion is that some Ca2+ activated process might damage the proteins involved in Ca2+ release.
This delayed recovery is likely to be an important cause of overtraining, which is seen in many sports. Many athletes and coaches believe that the more you train, the better you perform. However, there is a limit to the beneficial effects of training. With too much training the slow phase of recovery is never completed and performance can start to decline. Some athletes respond to this decline by training even more and a vicious cycle develops. Nevertheless, there is wide recognition of this problem and it is one of the factors which underlies the practise of ‘tapering’ of training adopted by most athletes before big events.
In conclusion, the answer to the question ‘What causes muscle fatigue?’ is ‘It depends on the type of activity’. While this answer is hardly inspiring during a dinner conversation, it is perfectly logical considering the numerous parallel processes that operate close to their maximum during intense muscle contractions.
References
Allen, D. G. & Westerblad, H. (2001). Role of phosphate and calcium stores in muscle fatigue. J Physiol 536, 657-665.
Merton, P. A. (1954). Voluntary strength and fatigue. J.Physiol.(Lond.) 123, 553-564.
Stephenson, D. G., Nguyen, L. T., & Stephenson, G. M. M. (1999). Glycogen content and excitation-contraction coupling in mechanically skinned muscle fibres of the cane toad. Journal of Physiology 519, 177-187.
Westerblad, H., Allen, D. G., & Lannergren, J. (2002). Muscle fatigue: lactic acid or inorganic phosphate the major cause? News Physiol Sci. 17, 17-21.
Westerblad, H., Bruton, J. D., Allen, D. G., & Lannergren, J. (2000). Functional significance of Ca2+ in long-lasting fatigue of skeletal muscle. Eur.J Appl.Physiol 83, 166-174.
Westerblad, H., Lee, J. A., Lamb, A. G., Bolsover, S. R., & Allen, D. G. (1990). Spatial gradients of intracellular calcium in skeletal muscle during fatigue. Pflügers Archiv European Journal of Physiology 415, 734-740.
Westerblad, H., Lee, J. A., Lännergren, J., & Allen, D. G. (1991). Cellular mechanisms of fatigue in skeletal muscle. American Journal of Physiology 261, C195-209.
