
Physiology News Magazine

Muscular dystrophy and the brain
Features
Muscular dystrophy and the brain
Features
Stewart I Head (1) & John W Morley (1,2)
1: School of Medical Sciences, University of New South Wales
2: School of Medicine, University of Western Sydney, NSW, Australia
https://doi.org/10.36866/pn.68.34

Duchenne muscular dystrophy (DMD) is the second most common fatal genetic disease of human beings. It occurs with an incidence of about one in every 3500 live male births. The main characteristic of the disease is a progressive loss of skeletal muscle tissue, which eventually leads to the death of the afflicted boy. DMD was first described in the 17th century by the French physician Duchenne de Boulogne, and also in England by the British physician Edward Meryon. In his first description of the disease, Duchenne noted the boys often showed significant mental impairment. Studies in the latter half of the 20th century confirmed the mental impairment, with DMD boys having an average IQ of 85, and in addition, independently of their actual IQs, DMD boys have a poor performance in digit span, story recall, comprehension, serial position memory, reading and mathematical ability (Anderson et al. 2002). In boys, perhaps because of the severity of the skeletal muscle wasting of the disease and individual variability in the alterations of brain function, possibly reflecting the diversity of dystrophin gene mutation, the cognitive impairment has largely been overlooked. The mdx mouse, the most commonly used animal model of muscular dystrophy, also exhibits a range of behavioural and cognitive deficits (Anderson et al. 2002).
Dystrophin localization studies have identified at least seven isoforms of dystrophin, each with its own tissue-specific promoter, of which four are found in the CNS (Kunkel et al. 1985). Dystrophin is not uniformly distributed throughout the CNS, work on the mdx mouse shows that it is found in discrete areas of the brain and associated with specific neurons (Górecki & Barnard 1997). Clusters of dystrophin are most abundant on the soma and proximal dendrites of the pyramidal cells in cerebral cortex and hippocampus and on cerebellar Purkinje cells (Fig. 1A). In contrast to skeletal muscle, where dystrophin is distributed uniformly on the inside of the membrane, in neurons dystrophin is found in discrete punctate units localised at the postsynaptic density. Intriguingly, punctate clusters of dystrophin are not found at all postsynaptic densities, rather they are extensively co-localised with the GABAA inhibitory receptor in cerebral cortex, hippocampus and cerebellar Purkinje cells.
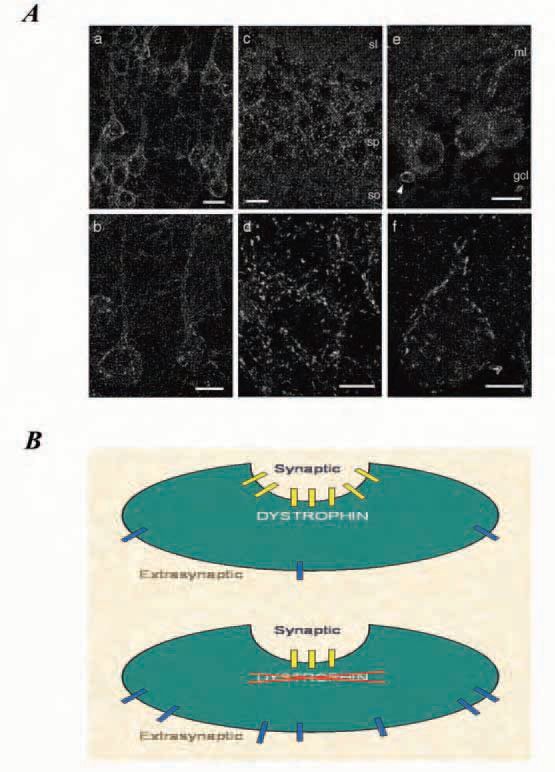
Knuesel and colleagues (1999) elegantly demonstrated that in mdx mice the absence of dystrophin from the postsynaptic clusters does not eliminate clustering of the GABAA receptors, rather, it reduces both the number and size of clusters at the postsynaptic density of the inhibitory synapses. In our laboratory we used Western blots to show that there is no difference between littermate control and mdx mice in the overall expression of GABAA receptor α1 subunit, which provides strong support for the suggestion that it is the clustering of receptors at the synapse that is affected in DMD, and that dystrophin may aid in trapping ion channels at the synaptic membrane and to aggregate them into large clusters for effective synaptic function. As a result, in dystrophic Purkinje cells it is likely that there is a substantially larger population of extrasynaptic GABAA receptors than in normal cells (Fig. 1B).
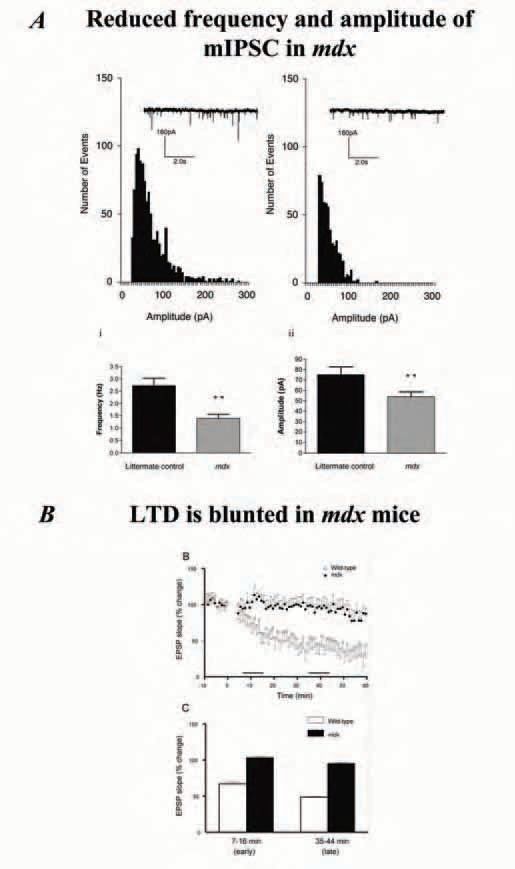
What role is dystrophin playing in the central nervous system? In skeletal muscle, it has been generally accepted that dystrophin plays an important role in maintaining normal [Ca2+ ]i and, more controversially, has a mechanical function in stabilising the fragile lipid bilayer against stresses associated with skeletal muscle contraction. It seems unlikely that dystrophin has a similar mechanical role in neurons, however, its absence may alter [Ca2+]i in neurons. As indicated above, in the CNS dystrophin is involved in the aggregation of large clusters of GABAA receptors at the postsynaptic inhibitory junction. In our laboratory we decided to use cerebellar brain slices from the mdx mouse to investigate the electrophysiological consequences of an absence of dystrophin from the Purkinje cell. The decision to use cerebellar Purkinje cells was an obvious one, primarily because Purkinje cells are the only neuron type in the cerebellum which express dystrophin and secondly because the Purkinje cells are large and easy to identify using infrared microscopy with differential interference contrast. Whole-cell patch clamping showed spontaneous miniature inhibitory post synaptic currents (mIPSCs) are reduced in frequency and amplitude in the dystrophic mice (Fig. 2A), a fairly predictable outcome of the reported reduction in the cluster size of the GABAA receptors. When we looked at an experimental model of synaptic plasticity, the effect of dystrophins absence was far from predictable. The Purkinje cell/parallel fibre synapse is a well studied model of long term depression (LTD). Using standard LTD induction paradigms we stimulated the parallel fibres in high-frequency bursts, while periodically mimicking the climbing fibre input to the Purkinje cell by depolarising the cell via a sharp intracellular microelectode. We deliberately chose to use the more classical sharp microelectrodes to record LTD in the Purkinje cells because we felt that if the absence of dystrophin altered the Ca2+ homoeostasis, this effect would be masked if the Purkinje cells were effectively [Ca2+]i clamped by dialysing them with a whole-cell patch clamp electrode. To our surprise we found a marked blunting of the LTD in the dystrophic Purkinje cells (Fig2B). Our finding of altered long-term synaptic plasticity in the mouse model of DMD is supported by work on a dystroglycan protein knockout model of congenital muscular dystrophy which showed blunting of hippocampal long term potentiation (LTP) (Moore et al. 2002).
At first sight the blunting of LTD in the cerebellum of the dystrophin-deficient mdx mice seems somewhat paradox-ical, given LTD involves the excitatory AMPA receptors and dystrophin is associated with the inhibitory GABAA receptors. However, when we consider that dystrophin may be playing a role in [Ca2+]i homoeostasis in the neurons, a pathological alteration of [Ca2+]i homoeostasis will alter induction of LTD as [Ca2+]i levels are critical in setting the thresholds for the induction of both LTD and LTP. We propose that one mechanism by which the[Ca2+]i homoeostasis could be disrupted in the Purkinje neuron by the absence of dystrophin is if the larger population of extrasynaptic GABAA receptors alters the influx of Ca2+. Fig. 1B illustrates what happens to the GABAA receptors when dystrophin is absent. We are currently in the process of using calcium sensitive dyes to test the extent to which [Ca2+]i levels are disrupted in the mdx Purkinje cells.
Whatever the underlying mechanisms, a change in the electrophysiological characteristics of GABAA inhibitory receptors and the blunting of long term synaptic plasticity in dystrophin-deficient muscular dystrophy will lead to a disruption in motor control and motor learning, and may underlie some of the CNS problems reported in both in the mdx mice and boys with DMD.
Acknowledgement
The work in our laboratory is supported by the Muscular Dystrophy Association, USA.
References
Anderson JL, Head SI, Rae C & Morley JW (2002). Brain function in Duchenne muscular dystrophy Brain 125, 4-13.
Górecki DC & Barnard EA (1997). Expression of dystrophin complex in the brain. In Dystrophin gene, protein and cell biology, ed. Brown SC & Lucy JA. Cambridge University Press, Cambridge, UK.
Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM (1999). Altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice). Eur J Neurosci 11, 4457-4462.
Kunkel LM, Monaco AP, Middlesworth W, Ochs HD & Latt SA (1985). Specific cloning of DNA fragments absent from the DNA of a male patient with an X chromosome deletion. Proc Natl Acad Sci 82, 4778-4482.
Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T & Campbell KP (2002). Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 25, 418(6896):376-377.