
Physiology News Magazine

Nanoscopy and cardiovascular physiology
Advances in single molecule localisation
Features
Nanoscopy and cardiovascular physiology
Advances in single molecule localisation
Features
Carl Harrison
Living Systems Institute, University of Exeter, UK
https://doi.org/10.36866/pn.110.36
While microscopy observes down to the micron level, nanoscopy aims towards a 10-fold increase in resolving power. Steps towards this aim are reported here in studies on the cardiac muscle ryanodine receptor type 2. As the RyR2 seems to be central to both physiological function and is altered in pathology, it is of great interest to the experimentalist and modeller alike.

In the last few decades, functional techniques have moved towards the molecular scale, such as patch clamp electrophysiology, atomic force microscopy or fluorescence resonance energy transfer (FRET) imaging. One key advance for these techniques was the ability to observe single molecules, rather than relying on ensemble averaged data. For nearly 150 years, it was widely accepted that the maximum resolution of a light microscope would be limited by the very nature of light itself to ~200nm. Immunofluorescence’s specificity has allowed so many details to
be observed about protein distribution, whilst electron microscopy (EM) unveiled amazing ultra-structural details at the nano-scale. Now, powerful new light microscopy techniques collectively called super-resolution microscopy (or nanoscopy) are bridging this gap towards single molecule information. There are numerous forms of nanoscopy, but here we discuss recent advances in single molecule localisation microscopy (SMLM) applied to cardiomyocytes.
SMLM techniques such as STORM or PALM rely on high laser power intensities, reducing buffers and some fundamental physics of fluorescence. Fluorophores are typically excited by a certain excitation wavelength being absorbed. When the molecule relaxes back to its ground state, the energy is then released (with a slight red shift) in the form of a photon, and the molecule is able to then be excited again. It is this constant cycling that results in a stable fluorescent signal in fluorescence microscopy. This property can be manipulated in the presence of high laser intensities and certain buffers. These conditions preferentially shift the fluorophore into a rarer meta-stable excited, ‘dark state’, whilst the remaining few molecules cycle through the fluorescence cycle rapidly, resulting in a brief, bright, ‘blink’ of light. The net result is that from an entire field of stable fluorescence one sees individual molecules blinking as diffraction-limited spots. The sample is imaged over time, each individual blink can then be localised around 10x more accurately than the blink itself and a composite super-resolution image is created. By introducing specific optical components, the shape of the blinks can be manipulated to divulge z-position information, resulting in three-dimensional super-resolution images.
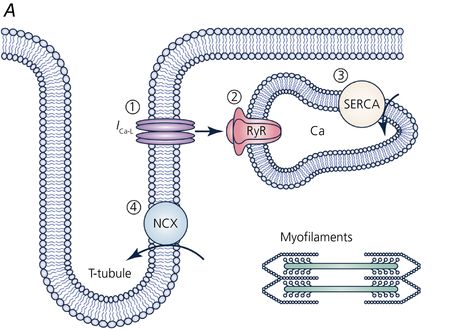
The study of the heart requires all the tools in physiology combining electrical, mechanical, biochemical, genomic, proteomic, and metabolomic aspects. Luckily, the cardiomyocytes that make up the muscular ventricular walls are fundamentally homogenous and so the study of isolated cardiomyocytes is considered to be approximately representative of the whole cardiomyocyte population. Within the cardiomyocyte, structures are highly ordered into longitudinal filaments for contraction and transverse membranous invaginations, known as t-tubules (first observed with EM). T-tubules facilitate the rapid distribution of electrical signals into the depths of the cell for the simultaneous activation of the muscle’s machinery. The tubules are directly opposed by internal organelles (sarcoplasmic reticulum; SR) and close to the contractile machinery. The currents flowing through ion channels in the t-tubules activate signalling pathways and internal ion channels such as the ryanodine receptor (RyR2) on the SR. RyR2 cluster activation results in quantal release of calcium, commonly observed as ‘calcium sparks’, and it is this calcium that activates the contractile machinery.
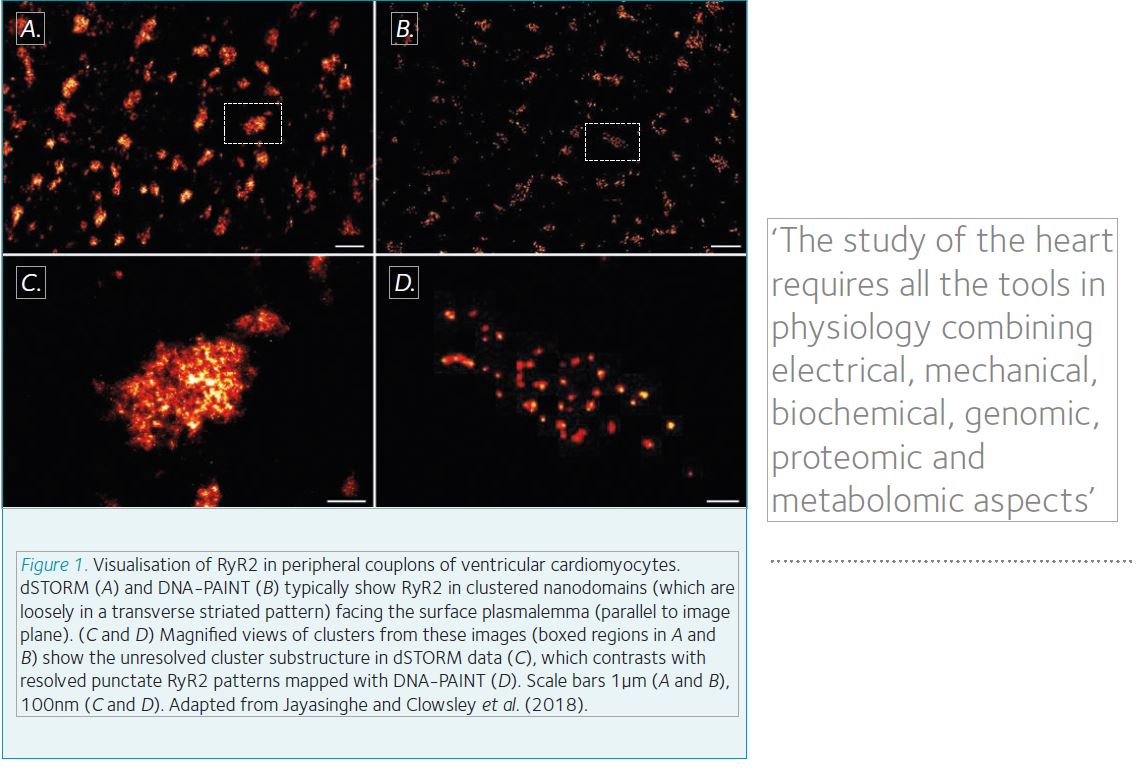
This nanodomain is where cardiomyocyte function is tightly regulated. Pathologies may arise if this orderly structure is perturbed, such as the loss of t-tubules in heart failure or arrhythmia. RyR2 are heavily implicated across a host of pathologies, and so there is great interest in their physiology. The Soeller lab at the University of Exeter has unveiled the spatial distribution of RyR2 at the nano scale since SMLM was demonstrated in biological samples. RyR2 clusters have been shown to follow a stochastic assembly with a near exponential size distribution. By utilising electrophysiological data and computer modelling, clusters within 100nm of each other can be thought of as superclusters. These act functionally as single calcium release units. By applying three-dimensional imaging, the lab has been able to estimate mean receptor capacities of ~63 RyR2 for the larger internal clusters and ~14 channels in the effectively flat peripheral clusters.
The apparent colocalisation of RyR2 with another SR protein, Caveolin3 was shown to be anti-correlated when seen at super-resolution. We demonstrated that another channel is involved in calcium handling, the sodium-calcium exchanger (NCX), and RyR2 showed a reduced colocalisation using super-resolution microscopy that would be indiscernible in diffraction-limited imaging, in a tamoxifen-induced junctophilin (JPH2) knockdown mouse. These findings, when taken together with findings that JPH2 knockdown mice exhibited an increased calcium spark frequency width and spark morphology, resulting in SR-calcium leakage, demonstrates how the use of fluorescence nanoscopy helps to explain findings in functional paired experiments.
An equal amount of our effort goes into tackling the technical challenges that come with imaging deep into optically thick tissues. Coupling a custom-built microscope with the development of PYME microscope environment software allows a great deal of technical control and flexibility. Our
lab has published on the considerations about different cameras, fluorophores and imaging buffers and how they stand up to biological imaging. Whilst it is not possible to discuss it here, it must be mentioned that other super-resolution techniques, such as stimulated emission depletion (STED) confocal imaging achieves similar resolution and results for RyR2. Structured illumination microscopy (SIM) or Airy Scan both provide 2x super-resolution and lend themselves to live cell imaging far more easily. These various techniques trade off speed, absolute resolution, technical difficulty and difficulty of live imaging.
More recently, we have been applying our expertise in imaging optically thick samples to other important physiological questions. We have collaborated on investigations into skeletal muscle physiology, the neuromuscular junction and neuronal cells. Nanoscopy is now starting to permeate through the biological and physiological journals and inform wider research. dSTORM microscopy has achieved protein cluster scale resolution, tantalisingly close to single molecule microscopy.
Our most recent work has allowed us to push into this regime by applying the DNA-PAINT method of staining pioneered by the Jungmann group at the Max Planck Institute of Biochemistry. The fundamental limits in dSTORM microscopy are the fluorescence capacity of the dye and finite number of these molecules. These properties are intrinsic to the fluorophore. DNA-PAINT uses antibodies that have a short, single-stranded DNA oligo attached to them. Then the complimentary oligo, with a fluorophore attached, is suspended free in the imaging media. At the right concentration of ‘imager strands’, they will stochastically bind to the ‘docking strands’. The length of the oligo dictates the binding time. By selecting an appropriate concentration of imagers with the right length, DNA-PAINT data resembles stochastic ‘blinks’ that we see in dSTORM. As the imagers are free in solution and can be replenished, they can be considered limitless, resulting in better resolution. Of equal power is the capability to perform multiplex imaging in series by using different sequences of oligos and wash steps. Crucially, this can be performed using the same fluorophore, reducing the optical complexity of the set-up and issues around chromatic aberrations.
These two powerful benefits to DNA-PAINT have allowed us to observe single RyR2 (a ~30 x 30 nm protein) channels within clusters for the first time at <10 nm resolution. We recently showed that RyR2 cluster morphology is dependent on JPH2 using dSTORM in JPH2 overexpression and tamoxifen-induced knockdown transgenic mice. JPH2 overexpression was associated with larger RyR2 clusters, but surprisingly these had a mild reduction in calcium release, consistent with a direct inhibition of RyR2 by JPH2. Our latest paper, published in Cell Reports, clearly shows that RyR2 receptors occupy the cluster areas observed in dSTORM images, with adequate space for accessory proteins such as JPH2 within the cluster. This intra-cluster detail will allow us to unravel the intricate architecture of these functional clusters. Hopefully, this will allow scientists to directly observe in situ molecular detail in health and disease.
References
Baddeley et al. (2009). Optical single channel resolution imaging of the Ryanodine receptor distribution in rat cardiac myocytes. PNAS 106(52), 22275-22280. DOI: 10.1073/pnas.0908971106
Eisner D (2017). Ups and downs of calcium in the heart. J Physiol 596(1), 19-30. DOI:10.1113/JP275130
Jayasinghe et al. (2018). True molecular scale visualization of variable clustering properties of ryanodine receptors. Cell Rep, 2(2), 557–567. DOI: 10.1016/j.celrep.2017.12.045
Jones et al. (2017). Cellular and molecular anatomy of the human neuromuscular junction. Cell Reports 21(9), 2348-2356. DOI:10.1016/j.celrep.2017.11.008
Jungmann et al. (2014). Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nature Methods. 11, 313–318. DOI: 10.1038/nprot.2017.024