
Physiology News Magazine

NO is pivotal in motor pathologies, but is it harmful or protective?
Dysregulation of the expression of nitric oxide synthase (NOS) is a hallmark of many neuropathological conditions. Using a model of peripheral motor neuropathy, we demonstrated that NOS up-regulation delays nerve repair and evokes many of the changes that occur in damaged motoneurons. Therefore, NOS might be a pivotal target for the development of therapeutic tools for diverse neuropathies.
Features
NO is pivotal in motor pathologies, but is it harmful or protective?
Dysregulation of the expression of nitric oxide synthase (NOS) is a hallmark of many neuropathological conditions. Using a model of peripheral motor neuropathy, we demonstrated that NOS up-regulation delays nerve repair and evokes many of the changes that occur in damaged motoneurons. Therefore, NOS might be a pivotal target for the development of therapeutic tools for diverse neuropathies.
Features
Bernardo Moreno-López
Grupo de Neurodegeneración y Neuroreparación (GRUNEDERE), Área de Fisiología, Facultad de Medicina, Universidad de Cádiz, Cádiz, Spain
https://doi.org/10.36866/pn.82.39

Traumatic injury of a peripheral motor nerve evokes physiopathological changes in the damaged motoneurons that are common to a broad array of neuropathological states, including dysregulation of protein expression, function and/or aggregation. One of the proteins involved is nitric oxide synthase (NOS), which synthesizes nitric oxide (NO), a highly reactive gas. First identified in biological systems as the endothelial-derived relaxing factor (EDRF), NO is an intercellular messenger that freely crosses plasma membrane, with multiple functions in the cardiovascular, immunological and nervous systems. The relative ubiquity of NO actions suggests that this molecule is important for physiological and pathological states and confers upon NOS important therapeutic possibilities for a variety of neuropathies.
There are three major isoforms of NOS, coded by different genes and differing in localization, regulation, catalytic properties and inhibitor sensitivity (Alderton et al. 2001). The isoform predominantly found in the neuronal tissue (nNOS) was the first to be purified and cloned. The inducible isoform (iNOS) occurs in a wide range of cells and tissues, including activated macrophages, and in pathological states of the central nervous system, in astroglia and microglia. Finally, the primary isoform found in vascular endothelial cells is known as eNOS. At the low tissue concentrations of NO generated by nNOS and eNOS, NO acts as a molecular messenger in multiple physiological transduction pathways; at high concentrations, such as those produced by iNOS, NO is cytotoxic. Remarkably, over-expression of eNOS or nNOS in pathological conditions can also yield toxic NO levels.
It is also noteworthy that NOS expression is up-regulated in motoneurons and reactive astrocytes in amyotrophic lateral sclerosis (ALS) and in neurons and/or glial cells in multiple sclerosis, HIV dementia, and Alzheimer’s, Parkinson’s and Huntington’s diseases, to cite the most prevalent neurological pathologies. Up-regulation of this enzyme also occurs after peripheral and central traumatic lesions of the nervous system. As a result, unmasking the role of NOS imbalance in neuron physiopathology is central to the development of feasible therapeutic tools. Over the past decade, our team has focused on investigating the role of NO in nerve repair and motoneuron pathology after the traumatic lesion of a nerve. We selected the hypoglossal motor system of the adult rat as an easily accessible system and crushing as the method of nerve injury.
Physical injury to a nerve is the most frequent cause of acquired peripheral neuropathy, which is responsible for loss of motor, sensory and/or autonomic functions. Injured axons in the peripheral nervous system maintain their capacity to regenerate in adult mammals. Therefore, the identification of molecules that regulate degenerative and regenerative processes is indispensable in developing therapeutic tools to accelerate and improve functional recovery following nerve injury.
This type of injury up-regulates the three major isoforms of NOS in the affected nerve: nNOS accumulates in the growing motor axons, eNOS is over-expressed in vasa nervorum in the distal stump and around the injury site, and iNOS is expressed by the recruited macrophages and phagocytic Schwann cells (Fig. 1).
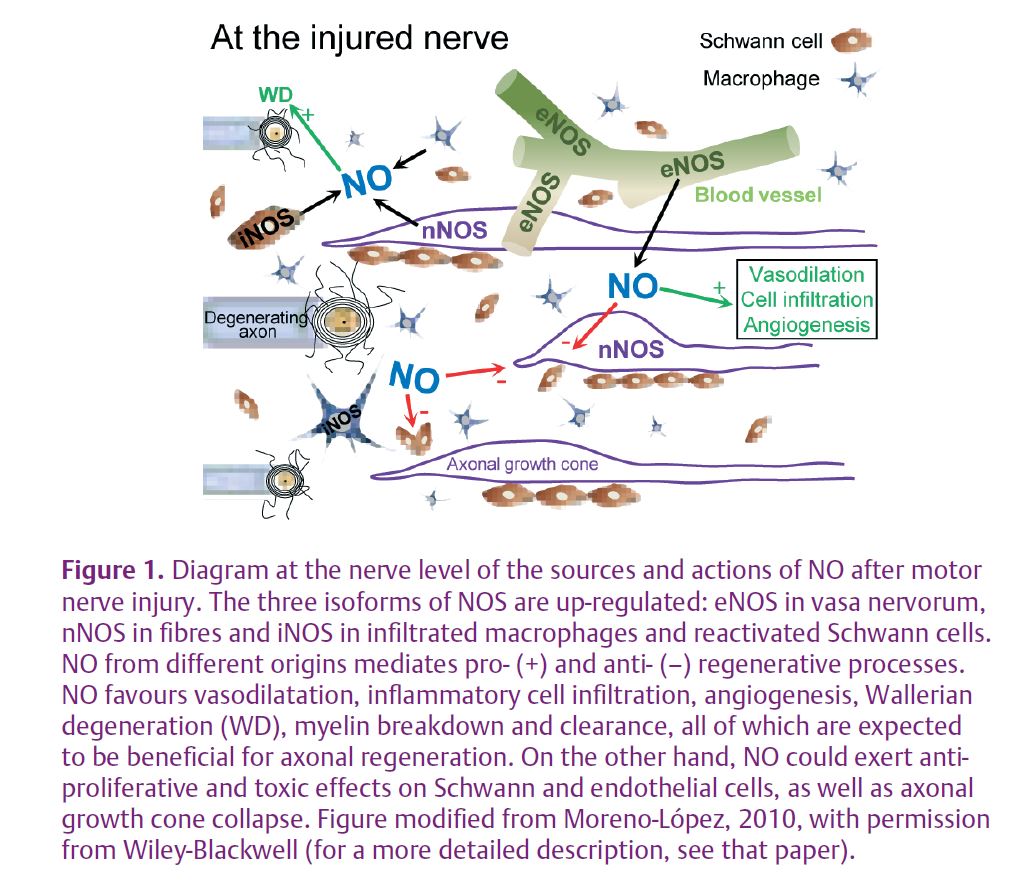
Although the pivotal role of NO is evident, whether it is harmful or protective is less clear. In nNOS or iNOS knockout mice, peripheral recovery is impaired due to delayed Wallerian degeneration, axon breakdown and Schwann cell reaction after nerve injury (Zochodne & Levy, 2005); this supports a beneficial role of NO. Paradoxically, chronic systemic inhibition of NOS accelerates the onset of functional muscle reinnervation, indicating a harmful impact on nerve repair because NO slows down axonal regrowth (Sunico et al. 2008). This apparent controversy was resolved by the finding that chronic, systemic, specific inhibition of eNOS – but not specific inhibition of n/iNOS – accelerated neuromuscular reconnection. To avoid unwanted side-effects due to the systemic administration of drugs and to improve specificity, an adenoviral vector was used to locally express a dominant negative of eNOS within the injured nerve. A single injection of this adenovirus on the day of nerve lesion increased axonal regeneration and significantly accelerated functional recovery of neuromuscular junction (Sunico et al. 2008). This suggests that NO of endothelial origin slows down muscle reinnervation by detrimental actions on axonal regeneration after peripheral nerve injury. These experiments identified eNOS as a potential therapeutic target for treatment of traumatic nerve injuries, highlighting the potential value of gene therapy using viral vectors to suppress the activity of eNOS.
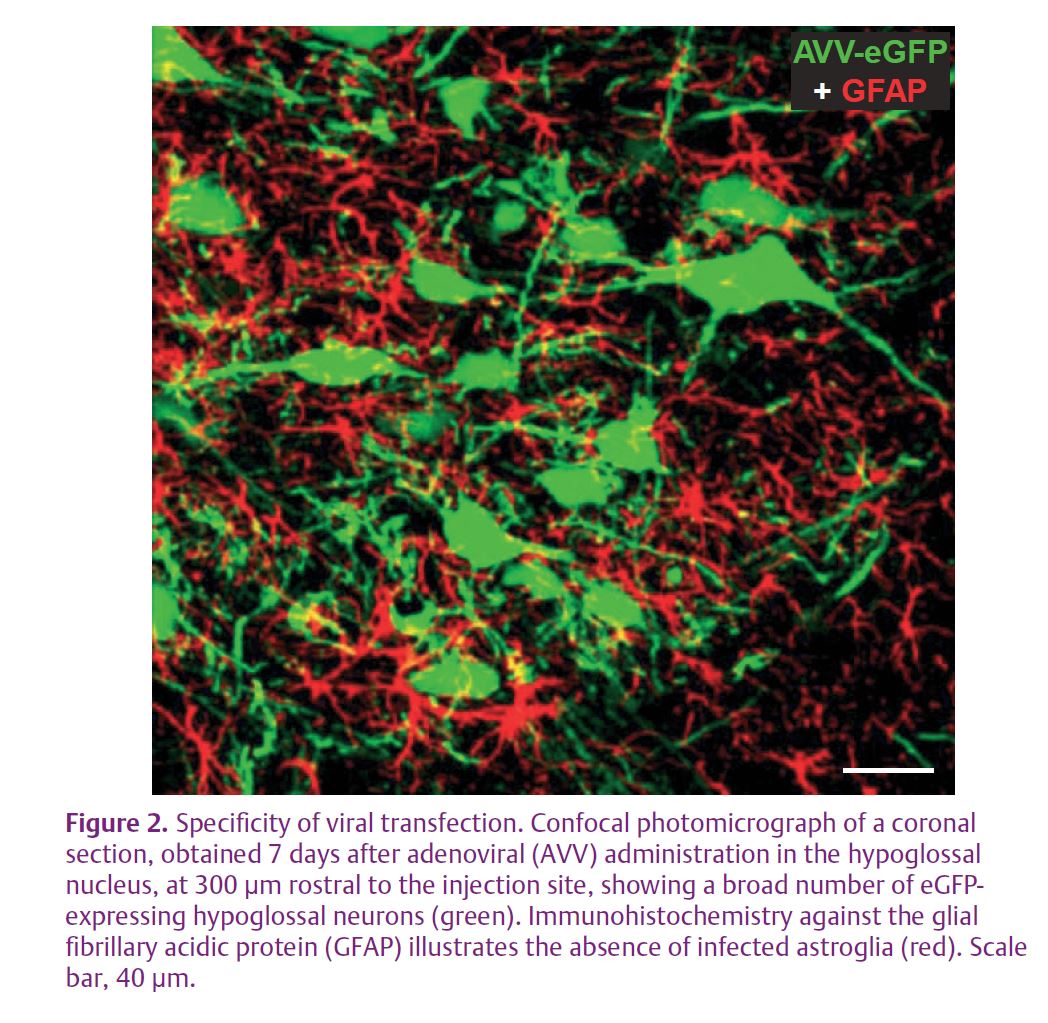
In a recent study reported in The Journal of Physiology, we analysed the role of induced NO in motoneuron pathology (Montero et al. 2010). To up-regulate nNOS in the hypoglossal motoneurons (HMNs), we used two different approaches. Crushing of the XIIth (hypoglossal) nerve provokes axonal transection of motoneurons. The alternative procedure was to transduce motoneurons by injecting an adenoviral vector, directing the expression of nNOS, into the hypoglossal nucleus (Fig. 2). The XIIth nerve crushing induces de novo nNOS expression in the perikarya of motoneurons, which normally lack this enzyme.
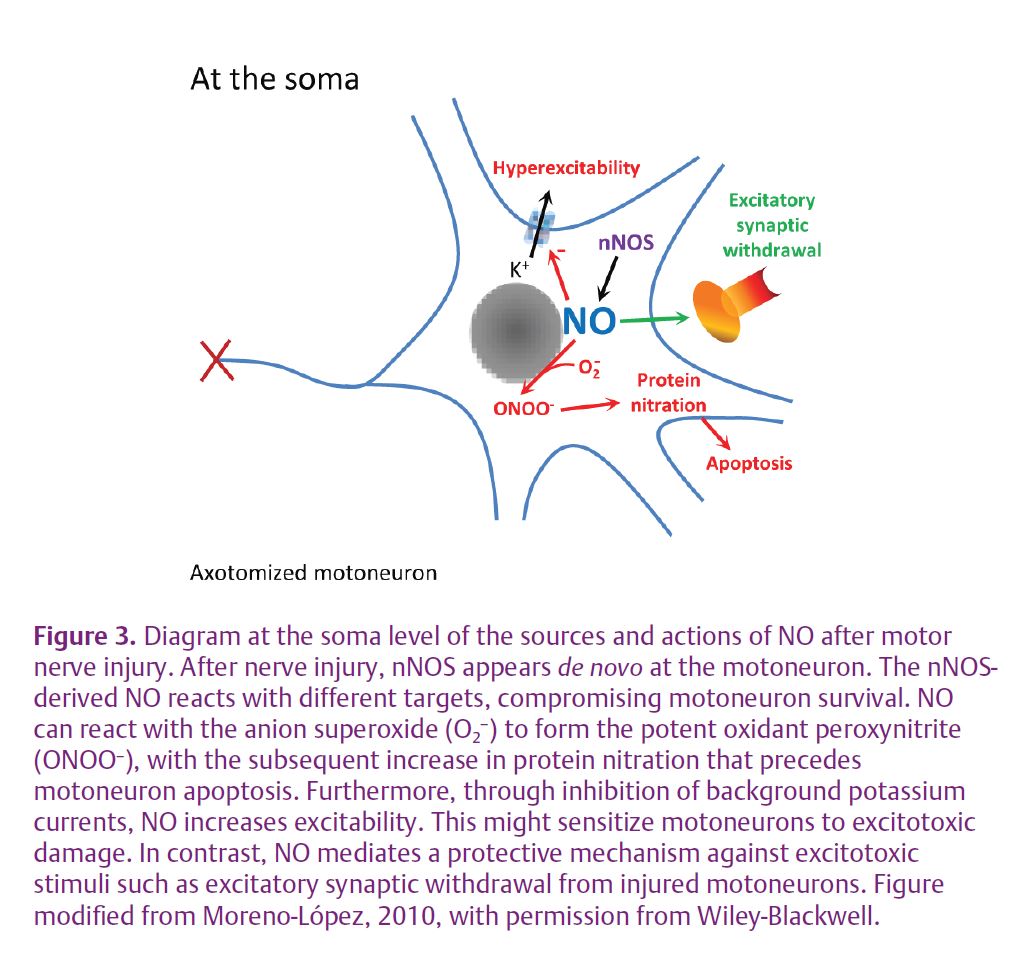
At the central level as well, NO has both harmful and beneficial roles for motoneuron survival after nerve injury. The gas is a pro-apoptotic molecule affecting motoneuron survival after motor nerve transection: nNOS deficiency or inhibition effectively reduced motoneuronal apoptosis induced by avulsion of the sciatic nerve in adult mice. Motoneuron apoptosis mediated by NO was accompanied by an increase in the NO subproduct peroxynitrite (ONOO–), a potent oxidant, and in protein nitration (Martin et al. 2005) (Fig. 3). In addition, chronic NO synthesis strongly increases intrinsic motoneuron excitability by inhibiting background potassium currents, suggesting that induced nNOS sensitizes injured motoneurons to apoptosis by mediating the hyperexcitability experienced by the motoneuron following axonal injury (Fig. 3). This NO-mediated action may sensitize motoneurons to excitotoxic damage (Gonzalez-Forero et al. 2007), with detrimental effects on motoneuron viability after axonal damage. On the contrary, however, nNOS up-regulation mediates the loss of excitatory synaptic inputs on injured HMNs (Fig. 3). This appears to be an NO-mediated protective action against excitotoxic stimuli. The activation of regrowth programs in adult motoneurons following axotomy may require a reversion to an immature electrical phenotype. Whether these apparently opposing impacts of NO on motoneuron survival form part of a motoneuron dedifferentiation process is a topic that merits attention.
Disruption of the trophic communication between moto -neurons and their target myocytes causes a well-characterized range of functional and synaptic impairments in insulted motoneurons. The NO-mediated disturbances involve changes in intrinsic membrane properties and anatomical synaptic deterioration that suggest a major pathological role of nNOS (Sunico et al. 2005, 2010; Gonzalez-Forero et al. 2007). However, nNOS is only one of the numerous proteins dysregulated after nerve damage. Thus, the actual role for nNOS is less clear within the complex synergistic and/or antagonistic actions of multiple dysregulated proteins. We further scrutinized the effects of virally induced de novo expression of nNOS in motoneurons, together with complementary attempts to down-regulate nNOS expression in damaged hypoglossal motoneurons using virally mediated gene knock-down (Montero et al. 2010). Our results demonstrate that nNOS over-expression in motoneurons is a sufficient signal to evoke many of the electrophysiological and anatomical changes associated with axonal damage.
These results point to nNOS as a pivotal target for the development of therapeutic tools for the treatment of peripheral neuropathies and neuro degenerative disorders characteristically accompanied by nNOS up-regulation. This finding also opens a line of research to establish a method for studying the role of dysregulated proteins in the neuronal impairment that occurs during the course of so many neuropathological conditions.
References
Alderton WK, Cooper CE & Knowles RG (2001). Nitric oxide synthases: structure, function and inhibition. Biochem J 357, 593–615.
Gonzalez-Forero D, Portillo F, Gomez L, Montero F, Kasparov S & Moreno-Lopez B (2007). Inhibition of resting potassium conductances by long-term activation of the NO/cGMP/protein kinase G pathway: A new mechanism regulating neuronal excitability. J Neurosci 27, 6302–6312.
Martin LJ, Chen K & Liu Z (2005). Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci 25, 6449–6459.
Montero F, Sunico CR, Liu B, Paton JF, Kasparov S & Moreno-Lopez B (2010). Transgenic neuronal nitric oxide synthase expression induces axotomy-like changes in adult motoneurons. J Physiol 588, 3425–3443. http://jp.physoc.org/content/588/18/3425.long
Moreno-Lopez B (2010). Local isoform-specific NOS inhibition: a promising approach to promote motor function recovery after nerve injury. J Neurosci Res 88, 1846–1857.
Sunico CR, Gonzalez-Forero D, Dominguez G, Garcia-Verdugo JM & Moreno-Lopez B (2010). Nitric oxide induces pathological synapse loss by a protein kinase G-, Rho kinase-dependent mechanism preceded by myosin light chain phosphorylation. J Neurosci 30, 973–984.
Sunico CR, Portillo F, Gonzalez-Forero D, Kasparov S & Moreno-Lopez B (2008). Evidence for a detrimental role of nitric oxide synthesized by endothelial nitric oxide synthase after peripheral nerve injury. Neuroscience 157, 40–51.
Sunico CR, Portillo F, Gonzalez-Forero D & Moreno-Lopez B (2005). Nitric oxide-directed synaptic remodeling in the adult mammal CNS. J Neurosci 25, 1448–1458.
Zochodne DW & Levy D (2005). Nitric oxide in damage, disease and repair of the peripheral nervous system. Cell Mol Biol (Noisy-le-grand) 51, 255–267.
Acknowledgements
This work was supported by grants from Spain’s Ministerio de Ciencia e Innovación (SAF2008-01415) and Consejería de Innovación, Ciencia y Empresa of the Junta de Andalucía (PAI2007-CTS-02606). I thank Elaine Lilly, PhD (Writer’s First Aid), for English language revision of this manuscript. I also thank Dr Sergey Kasparov’s laboratory (University of Bristol, UK) for collaboration with construction and gift of the viral vectors.