
Physiology News Magazine

NON-EXCITABLE CELL CALCIUM ENTRY – STATUS REPORT
Austin Elliott reports that, despite intensive research, there are still competing theories on how calcium entry in non-excitable cells is controlled
Features
NON-EXCITABLE CELL CALCIUM ENTRY – STATUS REPORT
Austin Elliott reports that, despite intensive research, there are still competing theories on how calcium entry in non-excitable cells is controlled
Features
Austin Elliott
University of Manchester, School of Biological Sciences, G38 Stepford Building, Oxford Road, Manchester M13 9PT
https://doi.org/10.36866/pn.44.5
“Stimulation of phospholipase C-coupled receptors in non-excitable cells causes an increase in intracellular free calcium ([Ca2+]i) consisting of two phases. The first phase is release of Ca2+ from intracellular stores, triggered by inositol 1,4,5-trisphosphate (IP3) acting on IP3 receptors (IP3Rs) on the ER, while the second phase is Ca2+ entry from the extracellular space”.
So far so good, and nowadays well-known Physiology undergraduate course material. Behind it, however, lies an interesting historical paradox. The importance of Ca2+ entry in controlling responses in cells other than muscles and nerves was actually recognised as long ago as the early 1960s (see Petersen, 1980, for some historical background), pre-dating the discovery of the Ca2+-releasing actions of inositol phosphates in 1983 by two decades. Another twenty years on, we now know rather a lot about how intracellular stores release Ca2+ (see review by Berridge, 1999). In contrast, the precise nature of the Ca2+ entry pathway, and the mechanism or mechanisms by which it is activated, continues to tantalise – and elude – researchers. Most work has concentrated on so-called store-operated or “capacitative” Ca2+ entry, in which the depletion of the intracellular stores somehow activates Ca2+ influx (Parekh & Penner, 1997). The question remains precisely how store depletion does this. A further complication is that there is still some debate as to whether this capacitative Ca2+ entry is the major, or indeed the physiological, pathway for stimulation-evoked Ca2+ influx.

What is the molecular identity of the Ca2+ entry channel?
So far the ion channel mediating depletion-activated Ca2+ entry has not been conclusively identified, despite considerable efforts. For most of the last decade homologues of a Drosophila protein, TRP, have been considered the most likely (only?) candidates. The trp gene was originally identified because mutations in the gene caused Drosophila photoreceptor cells to have grossly abnormal receptor potentials. On exposure to light, the photoreceptors from the mutant fly showed only a transient, rather than the normal sustained, depolarisation, since the Ca2+ entry that drives the sustained depolarisation was absent (for more details and references see Harteneck et al, 2000). Subsequently a family of mammalian TRPs have been identified and studied extensively in expression systems (Harteneck et al, 2000; Hofmann et al, 2000). Although it seems clear that over-expression of at least some of the TRP proteins can augment depletion-activated Ca2+ influx, none of the TRP work has shown convincingly that any TRP protein is the long-sought channel (Hofmann et al, 2000). Indeed, a paper published earlier this year in Nature argues that a different, though distantly related, Ca2+ channel protein, called CaT1, is the capacitative entry channel (Yue et al, 2001). The TRPs and CaT1 both share a number of structural features with other cation channel families, including Shaker-like voltage-dependent K+ channels (Figure 2). Many researchers working on TRPs believe that the TRPs may be components of multimeric Ca2+ channels, with different family members, and possibly other regulatory proteins, coming together to give a range of subtly different depletion- or messenger- gated channels. Similar logic will presumably apply to CaT1 and its homologues.
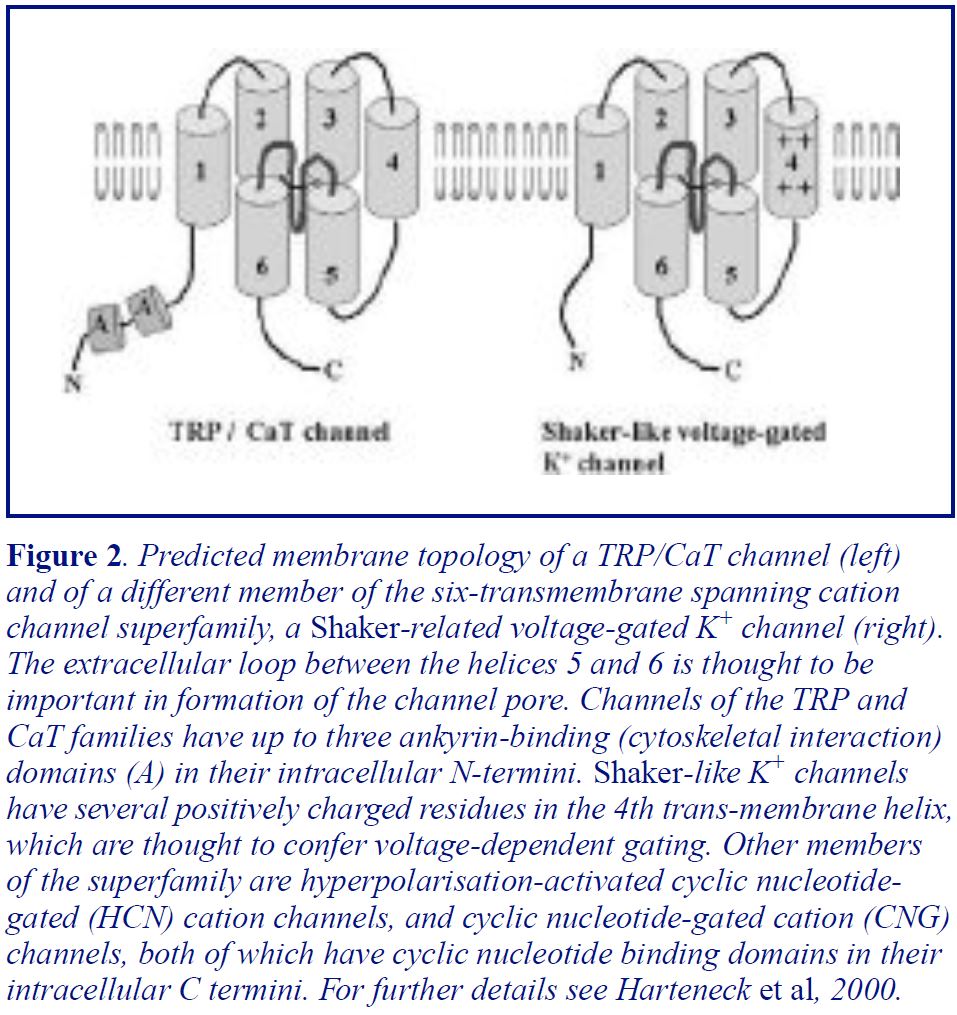
A family of depletion-activated Ca2+ entry channels might explain the fact that depletion- activated Ca2+ entry does not have identical properties in all cells, with differences being observed in cation selectivity/permeability (e.g. permeability to Mn2+ ions) and sometimes in sensitivity to blockers (for reviews see Parekh & Penner, 1997; Clementi & Meldolesi, 1996). The blockers most commonly used are divalent and trivalent metal cations such as Ni2+ and La3+. This highlights the important point that there is still no selective organic blocker of depletion- activated Ca2+ entry (for review see Clementi & Meldolesi, 1996). Given the importance of depletion-activated Ca2+ entry in regulating cellular functions (see below), selective inhibitors of the pathway could be expected to have many applications in medicine.
Who needs Ca2+ entry anyway?
Although the arguments continue over how Ca2+ enters non-excitable cells, there is no argument about the functional importance of Ca2+ entry. The essential role of TRP in Drosophila phototransduction provides one obvious example, but there are many others. In one intriguing study, Lewis and Crabtree used a clever genetic selection strategy to generate mutant lymphoma cells with defective depletion- activated Ca2+ entry. The mutant cells with impaired Ca2+ entry were unable to activate interleukin gene transcription properly in response to antigen presentation (Fanger et al, 1995). Many other responses in non-excitable cells which are driven by changes in [Ca2+]i are also inhibited by blocking Ca2+ influx. It should be noted, though, that this is not a rule – in some non-excitable cells the release of stored Ca2+ alone is enough to produce a full functional response. Indeed, in some non-excitable cells sustained Ca2+ entry may actually be associated with cell death (Raraty et al, 2000).
What triggers Ca2+ entry following store depletion?
There are two main theories as to how store depletion activates capacitative Ca2+ entry. The first theory suggests that store depletion produces a low molecular weight soluble messenger which diffuses to the plasma membrane to open the channels (Figure 3A). A wide range of candidates for this “Calcium Influx Factor” (CIF) have been suggested (for a comprehensive review of the earlier literature see the review by Barritt, 1999). None has stood the test of experiments in all systems, although work continues, with a recent paper arguing that a similar, though as yet unidentfied, molecule serves this function in yeast and in mammalian cells (Csutora et al, 1999). The second theory, which has gained ground as various candidate “CIFs” have been discredited, suggests that the IP3R itself interacts physically with the Ca2+ channel to trigger channel opening. This theory, originally proposed by Irvine (Irvine, 1990) and subsequently championed by him and by Berridge, obviously requires the Ca2+ stores to be within a few nanometres of the plasma membrane. This so-called “conformational coupling” model has received strong support from several key articles in the last two years (see Elliott, 2001). In particular, Muallem’s lab has demonstrated a direct interaction between Ca2+ entry channels and IP3Rs (or protein fragments of IP3Rs) in isolated patches, while evidence from several other labs highlights the role of the cytoskeleton in maintaining close contact between the Ca2+ stores and the channels. There are also studies that suggest that vesicle insertion into the membrane may be involved in the activation of Ca2+ entry (Figure 3B). More extensive discussion of this area, and references to the original articles, can be found in several recent reviews (Elliott, 2001; Putney, 1999; Rosado & Sage, 2000).
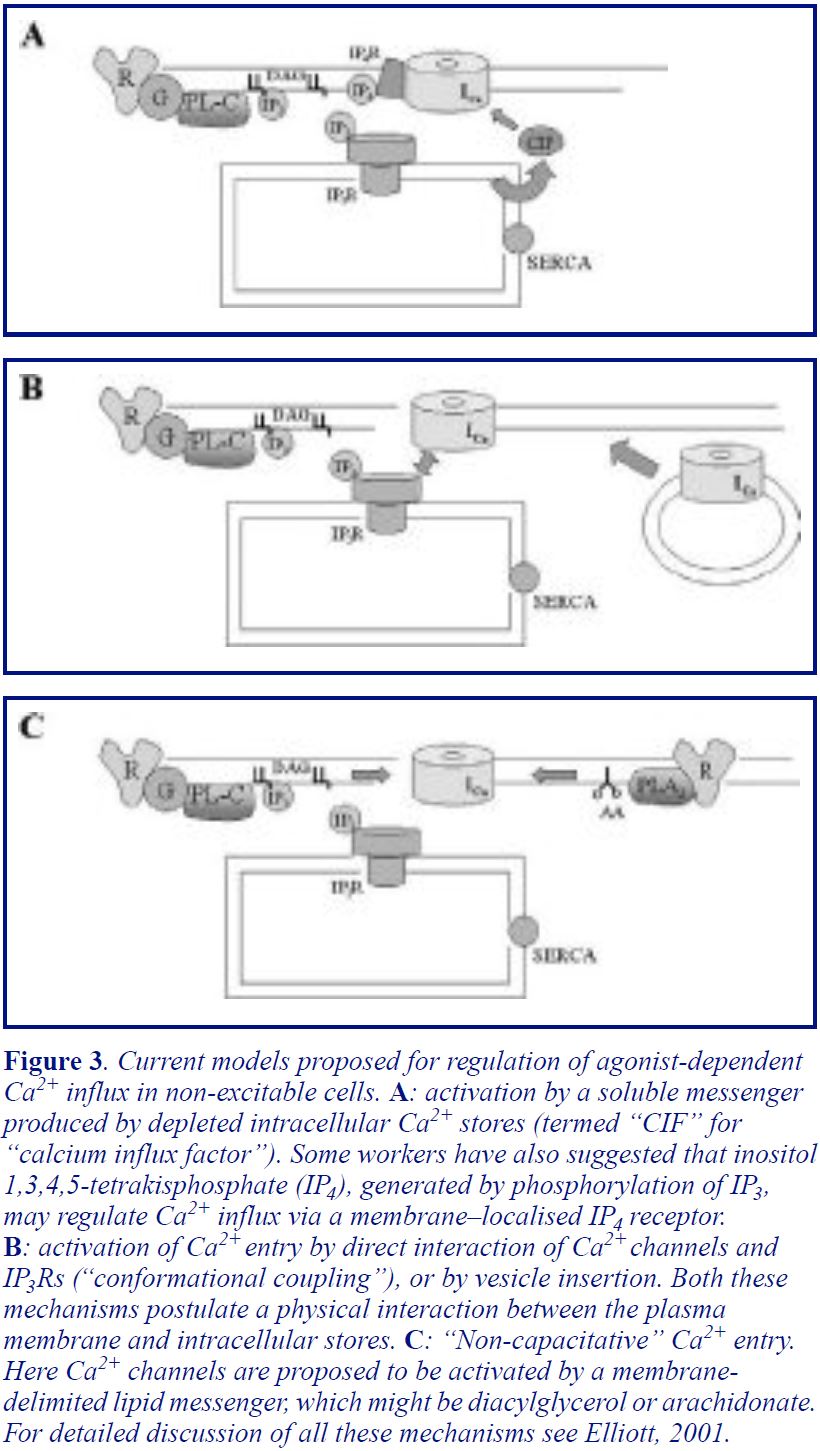
Is stimulation-activated Ca2+ entry necessarily capacitative?
Finally, there remains the possibility that capacitative Ca2+ entry may not represent the only, or even the critical, route for agonist-activated Ca2+ entry. For instance, there is good evidence that the closely related TRPs 3, 6 and 7 form a distinct subfamily of TRP channels which can be directly activated by diacylglycerol (Hofmann et al, 2000). These channels would therefore be activated following receptor stimulation but independent of store depletion. There is also evidence for non-capacitative Ca2+ entry activated by arachidonic acid, which is formed on stimulation of cells by activation of Phospholipase A2 (see Elliott, 2001 for refs). Shuttleworth has even argued that this arachidonate-activated non-capacitative pathway may be the dominant route of Ca2+ entry under physiological conditions (Shuttleworth, 1999).
Summary
Despite intensive research, there is no true consensus on the molecular identity, or regulation, of the depletion-activated Ca2+ entry pathway. Hopefully, further expression and cloning studies will confirm whether CaT1, TRP, or some other channel protein is the actual molecule underlying depletion-activated entry. In terms of regulation, conformational coupling models are currently in the ascendant over “CIF” theories, although the evidence for soluble messengers in some systems continues to accumulate. Finally, there remains lingering suspicion that depletion-activated entry may not necessarily be the physiological route for stimulation-evoked Ca2+ entry in many cells. Progress in all these areas can be expected as the study of non-excitable cell Ca2+ entry continues “beyond 2000”.
References
Petersen OH. (1980). Monographs of the Physiological Society No 36. Academic Press.
Berridge MJ. (1999). J Physiol 499: 291-306.
Parekh AB & Penner R (1997). Physiol Rev 77: 901-930.
Putney JW Jr. (1986). Cell Calcium 7: 1-12. Harteneck C, Plant TD, Schultz G. (2000). Trends Neurosci 23: 159-166.
Hofmann T, Schaefer M, Schultz G & Gudermann T. (2000). J Mol Med 78: 14-25.
Yue L, Peng J-B, Hediger MA & Clapham DE (2001). Nature 410: 705-709.
Clementi E & Meldolesi J. (1996). Cell Calcium 19: 269-279.
Fanger CM, Hoth M, Crabtree GR & Lewis RS. (1995). J Cell Biol 131: 655-67.
Raraty M, Ward J, Erdemli G, Vaillant C et al. (2000). PNAS USA 97: 13126-31.
Barritt GJ. (1999). Biochem J. 337: 153-159. Csutora P, Su Z, Kim HY, Bugrim A et al. (1999). PNAS USA 96: 121-126.
Irvine RF (1990). FEBS Lett. 263: 5-9.
Elliott AC. (2001). Cell Calcium 30: (In press).
Putney JW Jr. (1999). Cell 99: 5-8.
Rosado JA & Sage SO. (2000). J Physiol. 526: 221-9. Shuttleworth TJ. (1999). Cell Calcium 25: 237-246.