
Physiology News Magazine

Novel player in insulin resistance: focal adhesion kinase
Focal adhesion kinase (FAK) has recently been implicated in the regulation of insulin resistance in vitro. We hope the results of this study will provide a new strategy for the synthesis of new chemicals for FAK activation and promote development of new drugs against insulin resistance
Features
Novel player in insulin resistance: focal adhesion kinase
Focal adhesion kinase (FAK) has recently been implicated in the regulation of insulin resistance in vitro. We hope the results of this study will provide a new strategy for the synthesis of new chemicals for FAK activation and promote development of new drugs against insulin resistance
Features
Bharti Bisht (1,2), K Srinivasan (3), and Chinmoy S Dey (1)
1: Signal Transduction Research Laboratory, Department of Biotechnology, and 3: Department of Pharmacology and Toxicology, National Institute of Pharmaceutical Education and Research (NIPER) Sector 67, S.A.S. Nagar, Punjab 160 062, India
2: Current address: Department of Biology, Indian Institute of Science Education and Research (IISER), Mohali, Transit Campus: MGSIPAP Complex, Sector 26, Chandigarh 160 019, India
https://doi.org/10.36866/pn.75.17

Development of insulin resistance in the peripheral organs is one of the key defects associated with type 2 diabetes and obesity. However, the precise regulatory mechanisms of insulin resistance is not completely deciphered. Reports suggest cross-talk between focal adhesion kinase (FAK) and insulin signalling. Recent studies have associated FAK with insulin resistance. In vitro as well as in vivo studies have highlighted the role of FAK as a potential, novel player regulating insulin resistance. These studies have tremendous potential in presenting FAK as a possible drug target and in creating possibilities of new drug development against insulin resistance.
Insulin resistance and search for a novel target(s)
Insulin resistance is a condition in which normal concentrations of insulin produce a subnormal biological response in the primary target tissues such as skeletal muscle and adipose tissue (Taniguchi et al. 2006), and exhibit reduced insulin-stimulated glucose uptake and metabolism. Insulin resistance is a defect of insulin signal transduction. A variety of protein kinases and their substrates have been shown to be important regulators of insulin resistance at the molecular level. However, the signalling mechanisms involved remain elusive. Current estimates suggest that there will be over 350 million people worldwide affected by type 2 diabetes by the year 2030 (Turner & Heilbronn, 2008). Thus, considering the explosion of insulin resistance world wide there is an immediate need to explore novel and efficient drug targets.
Focal adhesion kinase (FAK) and insulin signalling
FAK is a non-receptor and non-membrane-associated protein tyrosine kinase found at the focal adhesions (Schlaepfer et al. 1999). FAK, a highly conserved protein expresses and undertakes a variety of functions in most tissues and cell types. Phosphorylated FAK can bind and integrate with multiple signalling pathways thereby acting as a molecular ‘switch’ in response to signals from the external environment and regulating various cellular processes (Romer et al. 2006).
In 1995, Knight et al. reported that insulin causes dephosphorylation of FAK, thus suggesting an antagonistic action of insulin on the integrin signalling. Co-immunoprecipitation of insulin receptor substrate-1 (IRS-1) with αvß3 integrin after insulin stimulation indicated a putative co-operation between two signalling proteins (Vuori & Ruoslahti, 1994). Czech and co-workers reported evidence for the crosstalk between integrin and insulin signalling molecules (Guilherme et al. 1998). Baron and her group have provided the first evidence demonstrating FAK as a direct substrate of insulin and a direct interplay between FAK and IRS-1; however, no interaction was observed between FAK and the insulin receptor (IR) (Lebrun et al. 1998). Further, it was observed that FAK could induce IRS-1 tyrosine phosphorylation resulting in an increased association of IRS-1 with p85α, SHP-2 and Grb 2. Lebrun et al. (2000) presented an interesting finding utilizing FAK-knockout fibroblasts where IRS-1 expression was observed to be abolished in FAK-knock out cells. Cheung et al. (2000) evaluated the role of FAK in TNF-α-induced insulin-resistant hepatocytes demonstrating that insulin acts as an activator of FAK by increasing its tyrosine phosphorylation. A new dimension was added to FAK functions when Huang et al. (2002) reported regulation of glycogen synthesis by FAK expression in hepatic cells. Data showed a direct interaction of FAK with GSK3-ß which made the group conclude that FAK acts downstream to IR, IRS-1 and PI3K via interaction with Akt and GSK3-ß. Recently the research group of Brayer-Ash knocked down FAK expression by 50% and reported impaired insulin-stimulated 2-deoxyglucose (2 DOG) uptake and concluded that the integrin signalling pathway potentially plays an important regulatory role in muscle insulin action (Huang et al. 2006). In light of the above findings, it is safe to conclude that FAK has important roles to play in regulating insulin signalling. However, questions regarding the physiological role of FAK in the regulation of insulin signalling, especially under pathophysiological conditions like insulin resistance, remain an enigma.
FAK and insulin resistance
In an effort to unwind the intertwined cascade of FAK, insulin signalling and insulin resistance, during 2007–2008 we have reported studies implicating FAK in the regulation of insulin resistance in vitro (Bisht et al. 2007; Bisht & Dey, 2008) and in vivo (Bisht et al. 2008), establishing the role of FAK in regulating insulin resistance.
In order to understand the molecular regulation of insulin resistance, we have developed insulin-resistant C2C12 skeletal muscle cells in our laboratory (Kumar & Dey, 2003). To understand the role of FAK, we approached the problem in two ways i.e. gain of function by overexpessing FAK and loss of function by silencing FAK. A significant decrease in tyrosine phosphorylation of FAK was observed in insulin-resistant C2C12 cells. Overexpression of FAK in insulin-resistant C2C12 skeletal muscle cells increased insulin sensitivity and glucose uptake (Bisht et al. 2007). These effects were reversed by expression of kinase activity mutant FAK or suppression of endogenous FAK by siRNA. FAK was also found to interact downstream of IRS-1, phosphatidylinositol 3-kinase (PI3K) and protein kinase C (PKC), leading to glucose transporter-4 (Glut-4) translocation and in up-regulation of glucose uptake. The study demonstrated for the first time, a direct role of FAK in insulin-resistant skeletal muscle cells. However, the underlying mechanism for FAK-mediated Glut-4 translocation leading to glucose uptake still remained unknown. We observed that overexpression of FAK induces actin remodelling and enhances co-localization of Glut-4 with actin, and thus elicits glucose uptake under insulin-resistant conditions (Bisht & Dey, 2008). FAK silencing by siRNA prevented actin remodelling, negatively affecting Glut-4 translocation and resulting in insulin resistance (Fig. 1). Further we observed that FAK regulates Glut-4 translocation via a PI3K-dependent pathway. Our study provided evidence that strongly supports the idea that modulation of FAK expression regulates insulin sensitivity in skeletal muscle cells in culture.
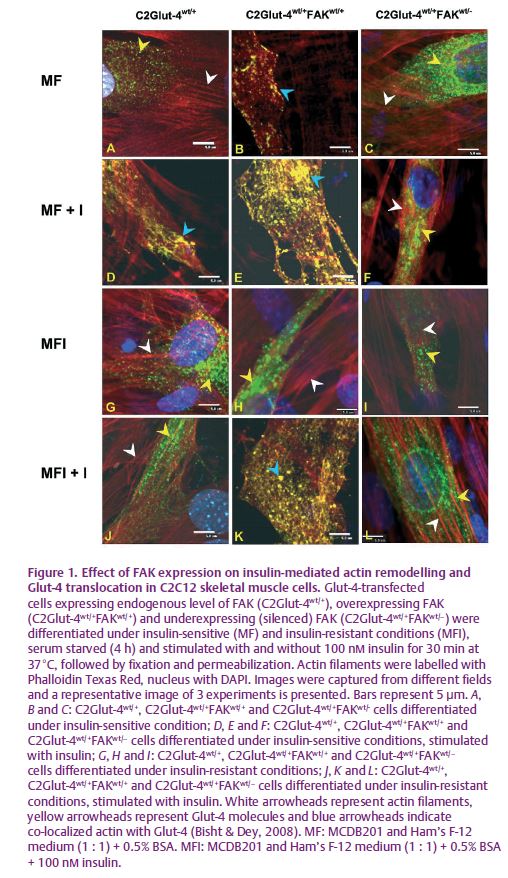
To prove conclusively the role of FAK in insulin resistance we down-regulated FAK expression using hydrodynamic tail vein injection of FAK-specific siRNA in mice in vivo (Bisht et al. 2008). We observed that FAK silencing impaired insulin signalling, altered glucose tolerance associated with weight gain and developed hyperglycaemia and hyperinsulinaemia (Fig. 2). FAK silencing resulted in inhibition of IRS-1 expression which in turn inhibited the downstream pathway leading to impaired insulin signalling. This provided direct and conclusive evidence that FAK is a crucial mediator of insulin resistance in vivo.
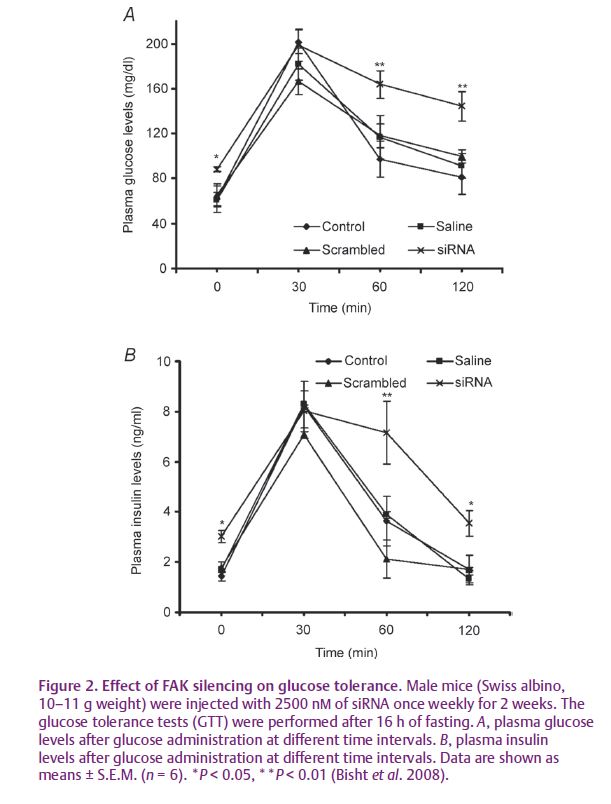
We believe our findings in vitro and in vivo will certainly be helpful in understanding the molecular basis of insulin resistance. Considering the lethality of the FAK gene knockout approach, our study will provide a new strategy for in vivo inhibition of FAK. Furthermore, the study will certainly motivate chemists to synthesize new chemical entities for FAK activation and promote development of new drugs against insulin resistance.
References
Bisht B & Dey CS (2008). Focal adhesion kinase contributes to insulin-induced actin reorganization into a mesh harboring glucose transporter-4 in insulin resistant skeletal muscle cells. BMC Cell Biol 9, 48.
Bisht B, Goel HL & Dey CS (2007). Focal adhesion kinase regulates insulin resistance in skeletal muscle. Diabetologia 50, 1058–1069.
Bisht B, Srinivasan K & Dey CS (2008). In vivo inhibition of focal adhesion kinase causes insulin resistance. J Physiol 586, 3825–3837.
Cheung AT, Wang J, Ree D, Kolls JK & Bryer-Ash M (2000). Tumor necrosis factor-α induces hepatic insulin resistance in obese zucker (fa/fa) rats via interaction of leukocyte antigen-related tyrosine phosphatases with focal adhesion kinase. Diabetes 49, 810–819.
Guilherme A, Torres K & Czech MP (1998). Cross-talk between insulin receptor and integrin signaling pathways. J Biol Chem 273, 22899–22903.
Huang D, Cheung AT, Parsons JT & Bryer-Ash M (2002). Focal adhesion kinase (FAK) regulates insulin-stimulated glycogen synthesis in hepatocytes. J Biol Chem 277, 18151–18160.
Huang D, Kohe M, Ilic D & Bryer-Ash M (2006). Reduced expression of focal adhesion kinase disrupts insulin action in skeletal muscle cells. Endocrinology 147, 3333–3343.
Knight JB, Yamauchi K & Pessin JE (1995). Divergent insulin and platelet-derived growth factor regulation of focal adhesion kinase (pp125fak) tyrosine phosphorylation, and rearrangement of actin stress fibers. J Biol Chem 270, 10199–10203.
Kumar N & Dey CS (2003). Development of insulin resistance and reversal by thiazolidinediones in C2C12 skeletal muscle cells. Biochem Pharmacol 65, 249–257.
Lebrun P, Baron V, Hauck CR, Schlaepfer DD & Van Obberghen E (2000). Cell adhesion and focal adhesion kinase regulates insulin receptor substrate-1 expression. J Biol Chem 275, 38371–38377.
Lebrun P, Mothe-Satney I, Delahaye L, Van Obberghen E & Baron V (1998). Insulin receptor substrate-1 as a signaling molecule for focal adhesion kinase pp125fak and pp60src. J Biol Chem 273, 32244–32253.
Romer LH, Birukov GK & Garcia Joe GN (2006). Focal adhesions paradigm for a signaling nexus. Circ Res 98, 606–616.
Schlaepfer DD, Hauck CR & Sieg DJ (1999). Signaling through focal adhesion kinase. Prog Biophys Mol Biol 71, 435–478.
Taniguchi CM, Emanuelli B & Khan CR (2006). Critical nodes in signaling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7, 85–96.
Turner N & Heilbronn LK (2008). Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol Metab 19, 324–330.
Vuori K & Ruoslahti E (1994) Association of insulin receptor substrate-1 with integrins. Science 266, 1576–1578.
Acknowledgment
This study was supported by a grant from the Department of Biotechnology, Government of India, New Delhi to C.S.D. (BT/HRD/34/04/2004; BT/PR3994/620 MED/14/498/2003). B.B. was a recipient of a Research Fellowship from the C.S.I.R, Government of India, New Delhi.