
Physiology News Magazine

Oxidative stress is harmful, and the TRPM2 channel bears part of the responsibility
Emerging evidence shows that oxidative stress can bring about harmful consequences via activating the TRPM2 ion channel.
Features
Oxidative stress is harmful, and the TRPM2 channel bears part of the responsibility
Emerging evidence shows that oxidative stress can bring about harmful consequences via activating the TRPM2 ion channel.
Features
Lin-Hua Jiang
School of Biomedical Sciences, Faculty of Biological Sciences, University of Leeds, UK
https://doi.org/10.36866/pn.108.36
It is well known that oxidative stress, arising from generation of excessive reactive oxygen species, weak or impaired antioxidant defence, or both, is a common factor in the pathogenesis of a variety of disease states. Recent studies reveal that the TRPM2 (transient receptor potential cation channel melastatin-related subfamily member 2) ion channel bears part of the responsibility for the harmful consequences of oxidative stress such as diabetes, post-ischaemia brain damage, and neurodegenerative diseases.

Generation of ROS and oxidative stress
For all mammals like humans, life relies on continuous access to oxygen (O2) because it is important for the efficient production of ATP, the cellular energy supplier. Molecular oxygen contains two unpaired electrons in its outer electron shell; this physiochemical property makes it readily available to generate a group of highly reactive chemicals called reactive oxygen species (ROS), such as the superoxide anion (O2−), hydrogen p.eroxide (H2O2), and the hydroxyl radical (OH). A biochemist can easily describe to us how O2− is generated from O2 by the electron transport chains in the process of producing ATP in mitochondria. We are also assured that cells are equipped with various enzymatic and non-enzymatic antioxidant means to eradicate ROS. For example, O2− can be converted to H2O2 by superoxide dismutases. Subsequently, H2O2 is changed to water by catalase or, in a bit more complicated scheme, H2O2 to .OH via the Fenton reaction and then to water. No harm done! This is almost true under healthy conditions, where ROS generation is well balanced with timely elimination. ROS at very low levels can be beneficial by serving as a physiological signalling molecule in maintaining normal tissue homeostasis via regulating cell proliferation, differentiation, and programmed cell death. Immune cells such as macrophage cells generate O2− in large quantities mainly via NADPH oxidases and use it as a weapon to kill pathogens through the process of phagocytosis. This is because ROS at high levels can modify nucleic acids, proteins, and lipids and impair their physiological functions. By the same mechanisms of action, high levels of ROS are harmful or dangerous to mammalian cells. Thus, accumulation of ROS, resulting from excessive generation, deficiency in intrinsic antioxidant defence, or both, leads to cellular oxidative stress. It has been well documented that oxidative stress is a critical factor in the pathogenesis of a variety of disease states. Emerging evidence supports the idea that the expression of the TRPM2 channel confers susceptibility to oxidative-stress-induced cell death, thereby bringing about harmful consequences such as diabetes, post-ischaemia or reperfusion brain damage, and Alzheimer’s disease.
A brief introduction: TRPM2 channel and its activation by oxidative stress

Mammalian TRPM2 (transient receptor potential melastatin-related subfamily member 2) is an integral membrane protein, belonging to the large family of transient receptor potential proteins. As illustrated in the insert of Fig. 1, the TRPM2 protein comprises six membrane-spanning segments and intracellularly residing amino and carboxyl termini. Four TRPM2 proteins interact to form a protein complex, in which the fifth and sixth membrane-spanning segments and the re-entrant loop connecting them from each of the four proteins or subunits come together to make a central aqueous ion-conducting pore. The TRPM2 channel is permeable to Ca2+ and other cations (Fig. 1) (Jiang et al., 2010). The C-terminal tail, highlighted in blue in the insert of Fig. 1, is similar in amino acid sequence to NUDT-9, an enzyme hydrolysing ADP-ribose (ADPR). This NUDT-9 homology domain provides a unique binding site for ADPR, and thereby allow specific gating of the TRPM2 channel by intracellular ADPR.
ADPR was long known as a by-product generated in the process of repairing ROS-induced DNA damage, engaging poly(ADPR) polymerase-1 (PARP-1) and p(ADPR) glycohydrolase (PARG) in the nucleus using nicotinamide adenine dinucleotide (NAD) as the ADPR donor (Fig. 1). ADPR is now recognised as a critical signalling molecule which selectively activates the TRPM2 channel. Excessive generation of ADPR occurs under oxidative stress. Therefore, interest has been rapidly escalating in the role that the TRPM2 channel plays in the pathogenesis of oxidative-stress-associated diseases.
TRPM2 channel mediates ROS-induced pancreatic β-cell death in diabetes
The β-cells represent the predominant type of cell in the islets of Langerhans of the pancreas. These cells are particularly important because they are responsible for making insulin and releasing it into the bloodstream in response to rising levels of glucose, and thereby help cells in the body convert sugar into energy. Loss of pancreatic β-cells leads to chronic diabetes, particularly type-1 diabetes. Type-1 diabetes arises from erroneous destruction of pancreatic β-cells by immune cells, resulting in deficient insulin production and severe hyperglycaemia, and causing life-threatening complications. Insulin deficiency due to pancreatic β-cell death also contributes to type-2 diabetes, the more common and complicated form of the disease. Pancreatic β-cells are weak in antioxidant defence and thus highly vulnerable to damage by oxidative stress. ROS-induced pancreatic β-cell death plays a critical role in the pathogenesis of diabetes.
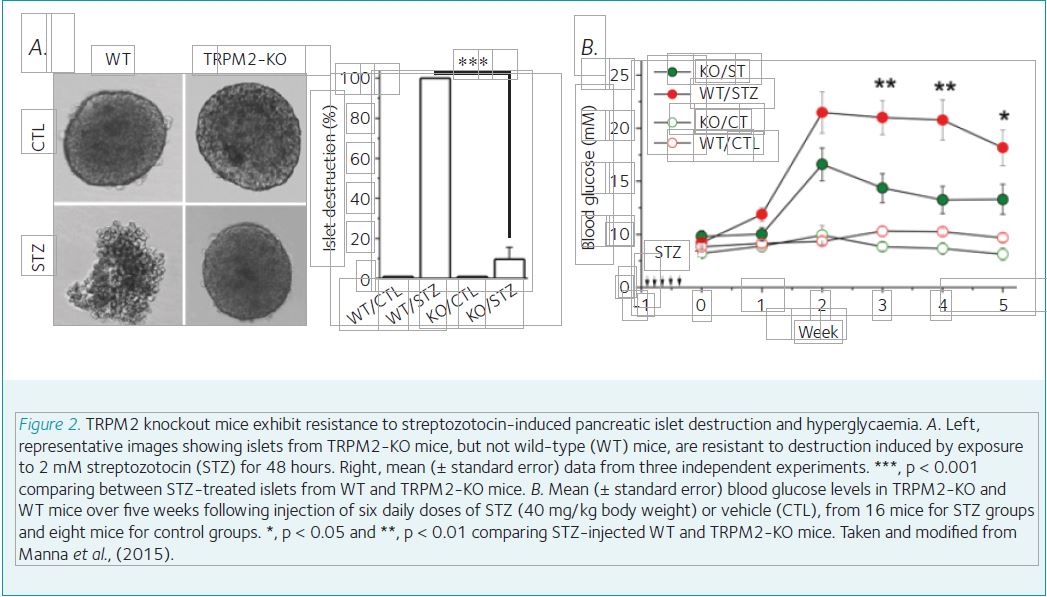
Many widely used diabetogenic agents such as streptozotocin bring about diabetes in rodent animals via inducing generation of excessive ROS. It was known that pancreatic β-cells and insulin-secreting insulinoma cells express a ROS-sensitive Ca2+-permeable channel and its activation is critically involved in ROS-induced cell death. This channel was later on identified as a TRPM2 channel. A recent study shows that genetic ablation of TRPM2 expression in mice provides strong protection against streptozotocin-induced destruction of pancreatic islets in vitro (Fig. 2a) and hyperglycaemia in vivo (Fig. 2b) (Manna et al., 2015). These findings reveal that the TRPM2 channel plays a critical role in oxidative-stress-induced pancreatic β-cell death and the pathogenesis of diabetes.
TRPM2 channel in delayed neuronal cell death and post-ischaemia brain damage
The brain constantly demands oxygen and glucose for energy production and is predisposed to damage as a result of loss of or reduction in blood supply such as ischaemic stroke. Ischaemic stroke represents one of the leading causes of adult mortality and morbidity worldwide. Ischaemia, if it is severe or long-lasting, causes brain damage and is life-threatening. The current mainstay therapeutic strategy for ischaemic stroke is to reinstate the blood circulation. Reperfusion following transient ischaemia can, however, be harmful because reperfusion provides O2 as the substrate for generation of excessive ROS, giving arise to oxidative stress and causing brain damage. The hippocampus is a critical brain region responsible for learning and memory. In both ischaemic stroke patients and rodent models of ischaemia-reperfusion brain damage, pyramidal neurons in the CA1 region of the hippocampus are highly susceptible to post-ischaemia brain damage. Such neuronal cell death often occurs with a substantial delay, and is often referred to as delayed neuronal cell death. Delayed neuronal cell death plays a vital role in post-ischaemia brain damage and cognitive dysfunction. Currently, there is no treatment for post-ischaemia brain damage.
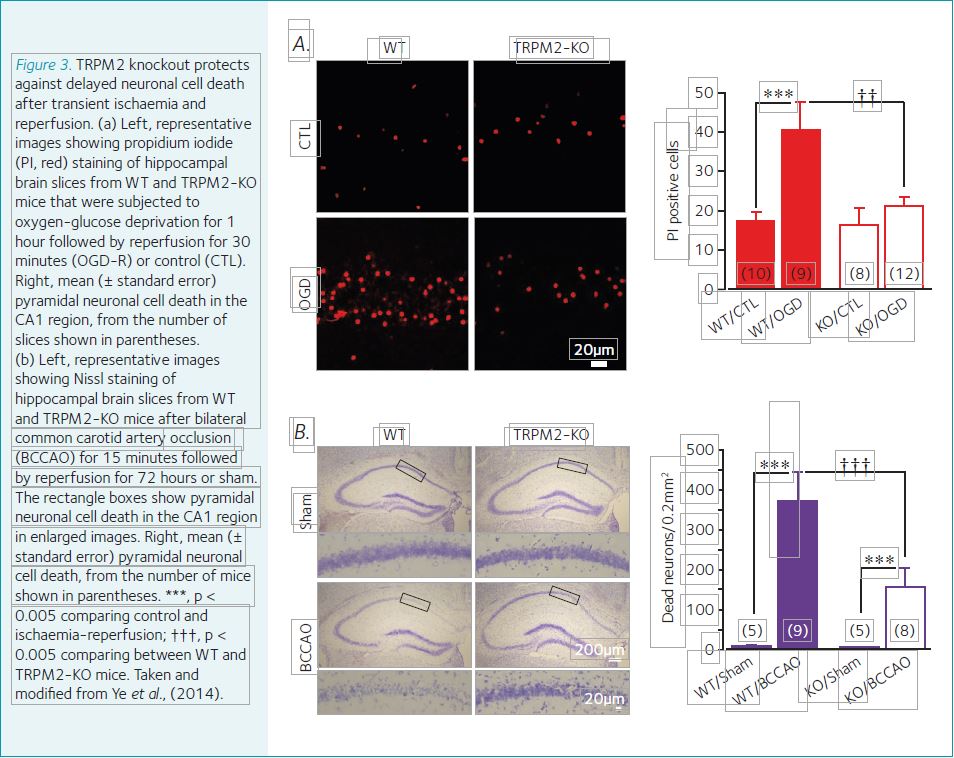
The TRPM2 channel is widely expressed in the brain, including in hippocampal pyramidal neuronal cells. Exposure to H2O2 activates the TRPM2 channel and results in hippocampal neuronal cell death. Genetic depletion of the TRPM2 expression in mice strongly prohibited delayed CA1 pyramidal neuronal cell death in vitro induced by oxygen-glucose deprivation and reperfusion (Fig. 3a) and brain damage in vivo after transient global ischaemia and reperfusion (Fig. 3b) (Ye et al., 2015). TRPM2 deficiency also effectively protected against post-ischaemia impairment in learning and memory (Ye et al., 2015).
These findings provide evidence to support a significant role for the TRPM2 channel in oxidative-stress-induced delayed neuronal cell death and post-ischaemia brain damage.
TRPM2 in amyloid β-induced neurotoxicity and Alzheimer’s disease pathogenesis
Alzheimer’s disease (AD) is a chronic neurodegenerative disease. It is the most prevalent cause of dementia among the elderly. One of the histopathological hallmarks is the formation of senile amyloid plaques containing high levels of neurotoxic amyloid β (Aβ) peptides. AD pathogenesis is highly complex, and the underlying mechanisms still remain a topic of ongoing debate. Extensive preclinical studies using AD rodent models and clinical analysis of human AD brains provide increasing evidence to support that Aβ-induced neurotoxicity and neuroinflammation are important in AD pathogenesis and progression. It is known that Aβ accumulation leads to cellular oxidative stress. Transgenic mice producing high levels of Aβ peptides exhibited greater microglial activation, neurotoxicity, synaptic loss, and age-related impairment in memory. In a recent study, it was shown that genetic deletion of the TRPM2 expression in this AD mice prevents all the above-mentioned Aβ overproduction-induced deleterious effects (Ostapchenko et al., 2015). This study reveals an important role for oxidative stress activation of the TRPM2 channel in Aβ-induced neurotoxicity and AD-related cognitive dysfunction.
In summary, there is increasing evidence from recent studies to support the idea that the TRPM2 channel plays a critical role in mediating oxidative-stress-induced pancreatic β-cell and neuronal cell death. Such TRPM2-mediated cell death contributes to the pathogenesis of diabetes, post-ischaemia brain damage, and AD. Therefore, the TRPM2 channel bears a significant part of the responsibility for the harmful consequences of oxidative stress.
References
Jiang LH et al. (2010). TRPM2 channel properties, functions and therapeutic potentials. Expert Opin Ther Targets 14, 973–88.
Manna PT et al. (2015). TRPM2-mediated intracellular Zn2+ release triggers pancreatic β-cell death. Biochem J 466, 537–46
Ye M et al. (2014). TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis 5, e1541
Ostapchenko VG et al. (2015). The transient receptor potential melastatin 2 (TRPM2) channel contributes to β-amyloid oligomer-related neurotoxicity and memory impairment. J Neurosci 35, 15157–15169