
Physiology News Magazine
Prometheus’ giblets
Chalones were at one time hypothetical factors that once secreted, controlled tissue regeneration and defined its ultimate size. Myostatin is just such a factor and although its ability to attenuate the growth of skeletal muscle is well known, recent studies suggest that it not only regulates cardiac muscle growth, but function as well
Features
Prometheus’ giblets
Chalones were at one time hypothetical factors that once secreted, controlled tissue regeneration and defined its ultimate size. Myostatin is just such a factor and although its ability to attenuate the growth of skeletal muscle is well known, recent studies suggest that it not only regulates cardiac muscle growth, but function as well
Features
Buel D Rodgers
Department of Animal Sciences and School of Molecular Biosciences, Washington State University, Pullman, WA 99164, USA
https://doi.org/10.36866/pn.78.19

Chained to a rock and forced to endure the eternal agony of having his liver ripped from his abdomen and devoured by a winged leviathan, Prometheus may have regretted mankind’s gift of fire against Zeus’ wishes. Being immortal, he endured this torture daily, for, as we all know, the liver regenerates. Ancient Greeks, by contrast, may not have known of chalones (pronounced ‘kay lones’, Greek for ‘to slacken’), secreted factors that inhibit tissue growth and control the regenerative process, in this case the liver’s, but they clearly understood that even titans have bad days.
The chalone–Prometheus association is discussed almost as often as the term chalone itself as both are commonly used to introduce the topic of liver regeneration. This process, however, isn’t limited to the liver per se as other tissues, skeletal muscle for example, also secrete factors that ultimately limit their growth. Indeed, myostatin is an extremely potent negative regulator of different growth processes that contribute to the size and functionality of skeletal muscle (Rodgers & Garikipati, 2008). The hypermuscularity of the myostatin null phenotype (Fig. 1) has been described in a variety of mammals, including humans, and is responsible for the ‘double muscling’ that occurs in some domestic breeds. The extreme nature of this phenotype, however, may have overshadowed other aspects of myostatin biology that until recently were overlooked.

Several lines of evidence suggest that myostatin may also regulate cardiac muscle (Rodgers & Garikipati, 2008). Its expression was identified in hearts of different mammalian, avian and fish species and it increases progressively as chick hearts develop. It also increases in some models of pathological cardiac hypertrophy. Myostatin inhibits proliferation and protein synthesis in a variety of cardiomyoblast cell culture models and reduces cardiac muscle mass when overexpressed in hearts of transgenic mice (Reisz-Porszasz et al. 2003). However, preliminary studies reported conflicting results on differences in heart size between wild-type and myostatin null mice (Rodgers & Garikipati, 2008), suggesting that null hearts could be larger or smaller, depending on sex or age, than wild-type hearts. A more thorough assessment of myostatin’s ability to regulate cardiac muscle growth and function was therefore needed.
We recently reported the most comprehensive analysis of myostatin null hearts to date the gestalt of which clearly indicates that myostatin is a negative regulator of physiological cardiac hypertrophy and, for the first time, excitation–contraction coupling (Rodgers et al. 2009). Myostatin inhibited basal and insulin-like growth factor (IGF)-stimulated cardiomyoblast proliferation in a dose-dependent manner. It also attenuated retinoic acid-stimulated differentiation while our cell culture system was found to express a full complement of myostatin receptors and binding proteins that matched mature cardiac muscle. Myostatin null hearts, normalized to tail length, were heavier than wild-type hearts at all ages and in both sexes (Fig. 2A). These results were confirmed by echocardiography, which also indicated that the larger heart mass was due to eccentric rather than concentric hypertrophy as the internal diameters and volumes were larger in myostatin null hearts despite similar wall thickness measurements (Fig. 2B). This is an important distinction as the compensatory response to large increases in skeletal muscle mass, which occurs in myostatin null mice, normally produces concentric hypertrophy. All of these results together indicate that myostatin directly and negatively regulates cardiac muscle growth. They also confirm previous studies with similar conclusions (Rodgers & Garikipati, 2008).
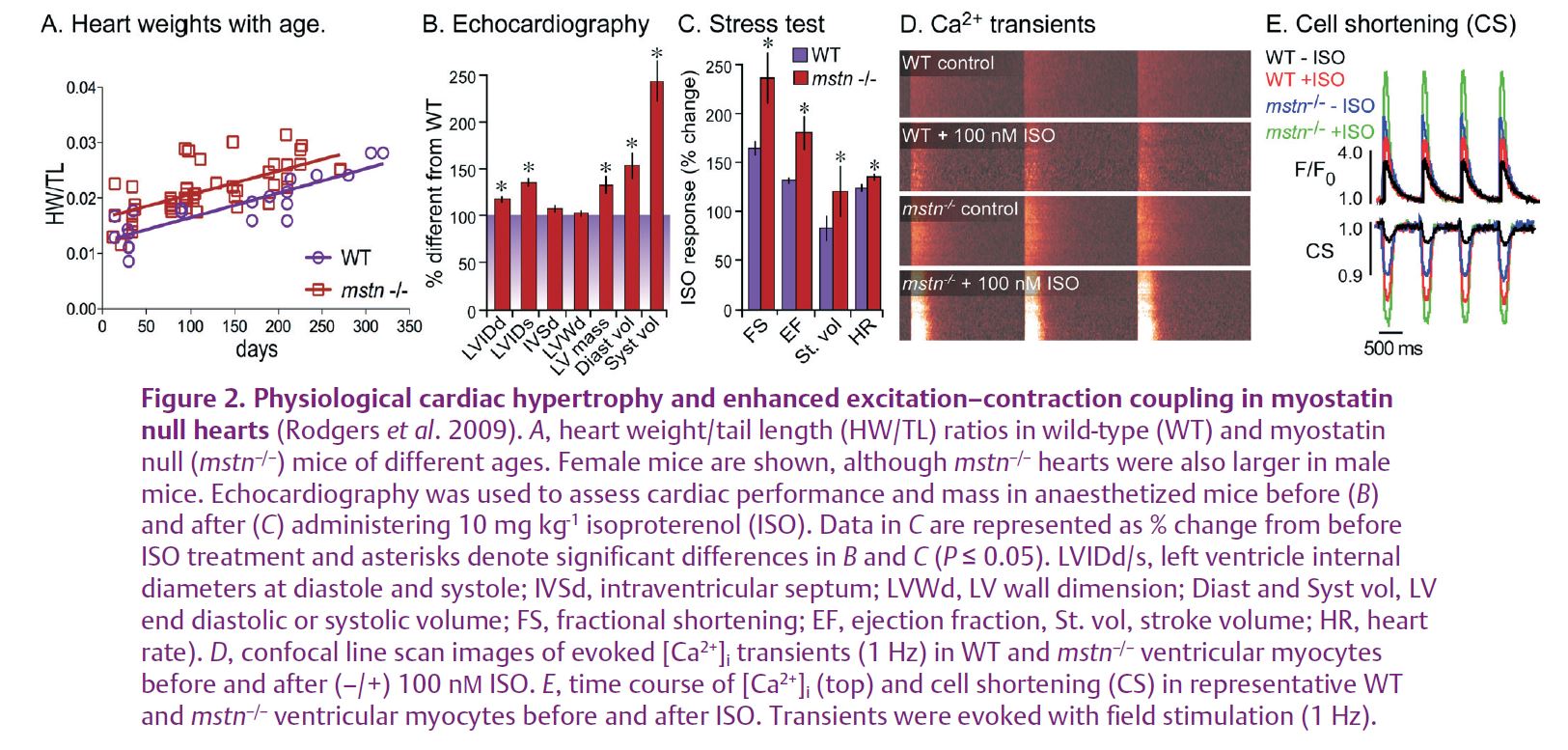
Most notably, we also discovered that myostatin null cardiac muscle was functionally superior to that of wild-type mice during an isoproterenol (ISO) stress test. In fact, changes in ISO-stimulated cardiac output, as indicated by several parameters (Fig. 2C), were significantly greater in myostatin null mice. This included heart rate and suggested that excitation–contraction coupling may also be enhanced. We confirmed this hypothesis in vitro using several biophysical assays and ultimately determined that [Ca2+]i transients (movements between intracellular stores) and total cellular loads were greater in primary ventricular myocytes from myostatin null hearts. Furthermore, these differences were associated with enhanced contractility in isolated cells (Fig. 2D and E). The superior β-adrenergic responsiveness and cardiac output in myostatin null hearts was therefore due in part to differences in Ca2+ handling and excitation–contraction coupling.
Normalized tension, ATPase activity and the tension–cost relationship of skinned fibres from wild-type and null hearts were identical and previous studies determined that the latter were histologically normal. We additionally determined that myostatin null hearts do not possess the fetal gene expression profile that occurs when hearts hypertrophy from pathological conditions, as with chronic hypertension or following myocardial infarction. Cardiac hypertrophy itself is not inherently bad as it is also an adaptive response to exercise. Myostatin null mice, therefore, possess physiological, not pathological, cardiac hypertrophy.
Myostatin-blocking technologies (e.g. immunoneutralization, soluble receptors, etc.) are currently being developed in both academic and industrial labs and all have produced very promising results (Rodgers & Garikipati, 2008). None, however, has thoroughly explored their potential in treating cardiac pathologies, and have focused primarily on muscular dystrophies and sarcopenia. Recent studies also suggest that attenuating myostatin action could potentially improve insulin sensitivity and reduce fat mass in obese subjects, most probably by increasing muscle mass and as a consequence, the resting energy expenditure (Guo et al. 2009). It is not unreasonable, therefore, to presume that such myostatin-blocking technologies may also help treat a variety of cardiac pathologies as well as two exacerbating conditions – obesity and type 2 diabetes.
The liver may be the most fabled regenerative organ. However, it is not alone as most organs possess at least a limited capacity to regenerate some or all of their tissues. This includes another giblet, the heart, whose growth is regulated by the chalone myostatin. One has to imagine, therefore, whether Prometheus would have suffered less had he lacked myostatin. He would have at least been in better shape to fight off that wretched bird.
References
Guo T, Jou W, Chanturiya T, Portas J, Gavrilova O & McPherron AC (2009). Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS One 4, e4937.
Reisz-Porszasz S, Bhasin S, Artaza JN, Shen R, Sinha-Hikim I, Hogue A, Fielder TJ & Gonzalez-Cadavid NF (2003). Lower skeletal muscle mass in male transgenic mice with muscle-specific overexpression of myostatin. Am J Physiol Endocrinol Metab 285, E876–E888.
Rodgers BD & Garikipati DK (2008). Clinical, agricultural, and evolutionary biology of myostatin: a comparative review. Endocr Rev 29, 513–534.
Rodgers BD, Interlichia JP, Garikipati DK, Mamidi R, Chandra M, Nelson OL, Murry CE & Santana LF (2009). Myostatin represses physiological hypertrophy of the heart and excitation–contraction coupling. J Physiol 587, 4873–4886. http://jp.physoc.org/content/587/20/4873.long