
Physiology News Magazine

Putting the brain into brainstem
What goes on in the brainstem and why should processes normally associated with higher parts of the brain be found much lower?
Features
Putting the brain into brainstem
What goes on in the brainstem and why should processes normally associated with higher parts of the brain be found much lower?
Features
Philip Winn
University of Strathclyde Glasgow, UK
https://doi.org/10.36866/pn.88.29
On the basis of experiments conducted in my lab we have concluded that brainstem functions are more sophisticated than previously thought, including critical processes of behavioural control. We argue that complex processes of learning and decision making are imperative for animals to thrive, and as such must be present in the oldest parts of brain – and we predict that when such processes go wrong, the results are catastrophic.
Visceral processes, respiration, arousal; semiautomatic skeletal functions, balance – familiar terms describing brainstem operations. In studying the pedunculopontine tegmental nucleus (PPTg), a small part of the mesopontine tegmentum, we have come to question whether these sorts of functions are enough to describe brainstem. The conventional view of PPTg is that it regulates locomotion and sleep, being described as part of the mesencephalic locomotor region and the ascending reticular activating system (Winn, 2008). However, in a series of experiments we have shown that rats bearing bilateral excitotoxic lesions of PPTg have deficits other than these. After recovery from surgery, lesioned rats have no impairment in locomotion, feeding, drinking, body weight maintenance or grooming. There is no dysfunction in normal sleep –REM or slow wave (Winn, 2008) – or in ‘emotional’ behaviour (Walker & Winn, 2007). But while lesioned rats seem normal, they actually have profound learning deficits.
The eight-arm radial maze (8ARM) tests spatial learning. For a random foraging task, rats were placed into the maze, with all eight arms open but only four baited with food – these four changed randomly trial by trial. Rats needed to find food without entering the same arm twice; the criterion for success was three consecutive days of testing with less than one error by the whole group. At the point when sham-operated rats achieved this, lesioned rats were at chance level, failing regardless of whether or not they were task-trained before surgery. However, lesioned rats’ latency for first arm entry, and their speed from arm to arm, was not different to controls: their failure to perform properly was explicitly not the product of motor deficit (Keating & Winn, 2002). In other experiments we examined intravenous self-administration (IVSA) of drugs. Rats were equipped with intra-jugular catheters connected to a syringe pump, operated by the rat pressing a lever. Two levers were available, but only one delivered amphetamine –pressing the other achieved nothing. Control rats steadily increased pressing on the reinforced lever over sessions: they like amphetamine. Lesioned rats did not learn and were no better on the last session than on the first. Two further experiments added to this: first, if rats were pre-trained to lever press for food (that is, they made a lever pressing–reward association before being lesioned) then pressing for amphetamine was exactly like that of controls. Despite the lesion, they pressed correctly and recognized the rewarding value of amphetamine. Second, regardless of prior training, lesioned rats did not perform well on a progressive ratio schedule of reinforcement where the number of lever presses per reward increased throughout the test (Alderson et al. 2004).
What do these studies tell us? Most clearly, there are no motor or motivational problems: lesioned rats moved freely around the 8ARM, ate food, lever pressed normally and liked amphetamine. Their deficit lies outside these domains.
The data can be accounted for as one of action–outcome association – understanding the relationships between actions and the outcomes produced. In the random foraging task, reward location varied on every trial, so rats had to remember where they had been and not go back – they needed to understand which actions led to which outcomes. Lesioned rats comprehensively failed and, because there was continual change in food location, pre-lesion training was worthless. In the IVSA task, rats were faced with two levers: pressing one did nothing, pressing the other gave drug. Again, lesioned rats could not associate action (correct lever pressing) and outcome (drug delivery) but in this test, if they had previously made an association between lever pressing in the same operant box and reward delivery – by pre-training to press for food – the presence of a bilateral PPTg lesion did not prevent expression of that association. But the progressive ratio schedule beat them – the relationship between action and outcome changed continuously, with more lever presses incrementally required for the next delivery of drug.
Two further things suggest that failure of action–outcome association is a viable hypothesis to explain the deficit: in recent experiments we examined contingency degradation, critical for identifying problems in action–outcome association; and examination of PPTg connections make a role in action–outcome association wholly plausible.
Contingency degradation asks whether, having learned an association, a rat can recognize that contingencies have changed so that they can stop responding. To do this we trained a group of rats on a random ratio–20 (RR20) schedule (0.05 probability of a lever press delivering a pellet; Fig. 1). Once they had all achieved consistent pressing rates we divided them into four groups: contingency degraded or remaining on RR20; with bilateral injections into PPTg of saline or muscimol, a GABA agonist that transiently inactivates it. Rats that were not contingency degraded maintained responding on RR20 regardless of whether they had saline or muscimol injected into PPTg: muscimol did not affect responding. When the contingency was degraded, rats receiving saline into PPTg reduced their lever pressing significantly: that is, they updated the action–outcome association, realizing that lever pressing was not delivering food. However, intra-PPTg muscimol led to rats maintaining high levels of lever pressing: there was no updating of the action–outcome association.
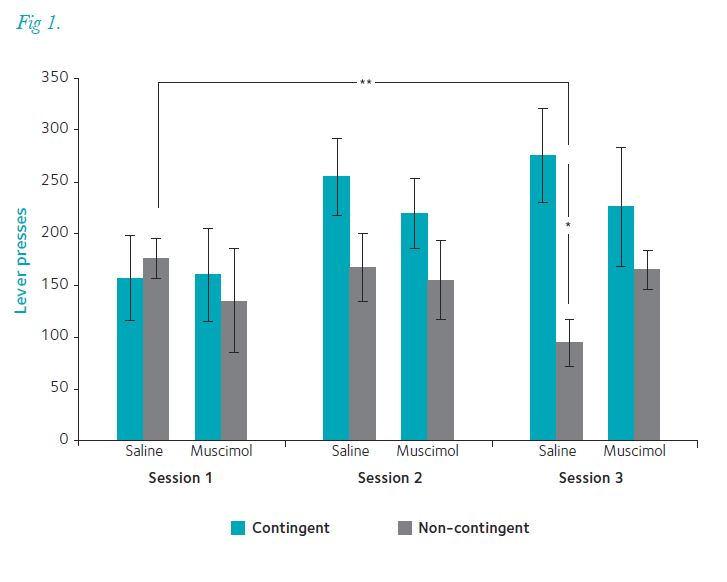
This all points to the conclusion that a functioning PPTg – transiently inhibited or permanently lesioned – is critical for forming and updating action–outcome association, a more sophisticated purpose for this level of brain than previously suspected. The anatomical connections of PPTg reinforce this (Fig. 2). Inputs descend from the forebrain – notably from basal ganglia output nuclei – and from sensory structures in midbrain and brainstem. (PPTg neurons show very fast responses to sensory input, especially auditory.) There are dense outputs to the thalamus – all thalamic nuclei have cholinergic input from the mesopontine tegmentum – and to sites of non-specific cortical input, such as the basal forebrain and lateral hypothalamus. Descending connections travel to the pontine reticular formation, medulla and spinal cord (Winn et al. 2009).

In the context of action–outcome association, however, the most significant connection is that with midbrain dopamine (DA) neurons (Maskos, 2008). These are known to be critical for learning, and their target sites in the striatum are involved in action–outcome association. Stimulation in the mesopontine tegmentum drives a three-phase release of DA in striatum: a fast spike of DA activity dependent on ventral tegmental area (VTA) nicotinic acetylcholine (ACh) and ionotropic glutamate receptors; decreased activity, mediated by muscarinic receptors at source in the brainstem; and a prolonged increase in DA release, dependent on VTA M5 ACh receptors (Lester et al. 2010).
What we suggest is that loss of PPTg input to DA neurons makes action–outcome association impossible because the normal regulation of DA neurons is dysfunctional.
We have argued that, as well as being a basal ganglia input/output station, PPTg also generates rapid responses to novel stimuli independently of forebrain systems (Winn et al. 2009; Wilson et al. 2009). It is part of a hierarchy of decision-making, operating on very short timescales so that animals can make immediate judgments about the need for action – avoidance of predators or capture of prey, for example.
PPTg anatomy is consistent with this: descending connections with motor and autonomic systems; ascending connections with basal ganglia (designed for action selection when there are competing response options); and inhibitory control of PPTg by forebrain systems, to prevent impulsive action. To make immediate judgments, it is critical that PPTg can recognize the significance of events. That it can do so is shown by primate electrophysiological data: one population of PPTg neurons responded proportionately to visual targets predictive of reward delivery and a different population to reward itself (Okada et al. 2009). This returns us to original notions of brainstem and its early-evolved functions, but with a critical difference. All vertebrates need to maintain basic physiological processes and control of these is naturally represented in older parts of the brain. But behavioural control is as important as physiological – animals that do the wrong thing don’t survive – so mechanisms of sensory analysis and decision-making must be represented in evolutionarily conserved older parts of the CNS (Wilson et al. 2009).
In the effort to understand the neural basis of psychological processing, neuroscience focuses on cerebral cortex and those allo- and sub-cortical structures intimately associated with it. However, because some behavioural decisions have to be made so quickly, key mechanisms of behavioural control must sit further down the neuraxis. What reach might these lower systems have into cognitive processing? It is a question worth asking, because it has been recognized for many years that midbrain DA neurons are involved in cognitive processing and in its disorders. Efforts to treat schizophrenia have focused almost exclusively on regulation of DA receptors in striatum and prefrontal cortex, in the belief that disturbed DA activity is the principal substrate of schizophrenia’s positive signs and symptoms (Frith, 1992). What if the disorder of DA neurons comes from a failure of control by structures that normally supply them with new information, as opposed to descending feedback control? The PPTg is known to be associated with endophenotypic features of schizophrenia, such as failure of prepulse inhibition and control of the auditory P300 as well as increased nicotine use. But more than these, it is conceivable that the catastrophic deficits we see in PPTg-lesioned rats when action–outcome association is needed reflect the problem that schizophrenic patients have in understanding their own and others’ actions – “patients misattribute self-generated actions to an external agent” (Frith, 1992, p.73). It is only speculation, but given the mismatch between the scale of impairment in schizophrenia and the failure to develop viable models of pathology or treatments, it is one worth making.
Critical to this speculation is something that we regard as secure: that brainstem systems have a sophisticated capacity to analyse incoming sensory data, understand that input in terms of what is already known and, if appropriate, make an immediate decision to act. It is the essential process used throughout the brain – analyse input, compare to experience, calculate the most appropriate response. The brainstem is just as brainy as the rest of it, but quicker.
Acknowledgements
I am grateful to many colleagues, including current and recent SIPBS lab members, Morag Farquhar, Nadine Gut, Katarzyna Pawlowicz, Amélie Gavard and Duncan MacLaren. This work has been funded by the Wellcome Trust, BBSRC and MRC.
References
Alderson HL, Latimer MP, Blaha CD, Phillips AG & Winn P (2004). An examination of d-amphetamine self-administration in pedunculopontine tegmental nucleus-lesioned rats. Neuroscience 125, 349–358
Frith CD (1992). The Cognitive Neuropsychology of Schizophrenia. Lawrence Erlbaum Associates, Hove UK.
Keating GL & Winn P (2002). Examination of the role of the pedunculopontine tegmental nucleus in radial maze tasks with or without a delay. Neuroscience 112, 687–696.
Lester DB, Rogers TD & Blaha CD (2010). Acetylcholine–dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci Ther 16, 137–162.
Maskos U (2008). The cholinergic mesopontine tegmentum is a relatively neglected nicotinic master modulator of the dopaminergic system: relevance to drugs of abuse and pathology. Br J Pharmacol 153, S438–S445.
Okada K, Toyama K, Inoue Y, Isa T & Kobayashi Y (2009). Different pedunculopontine tegmental neurons signal predicted and actual task rewards. J Neurosci 29, 4858–4870.
Walker SC & Winn P (2007). An assessment of the contributions of the pedunculopontine tegmental and cuneiform nuclei to anxiety and neophobia. Neuroscience 150, 273–290.
Wilson DIG, MacLaren DAA & Winn P (2009). On the relationships between the pedunculopontine tegmental nucleus, corticostriatal architecture and the medial reticular formation. In The Basal Ganglia IX, pp. 143–157, eds. H Groenewegen et al. Springer Science and Business Media, New York.
Winn P (2008). Experimental studies of pedunculopontine functions: are they motor, sensory or integrative? Parkinsonism Relat Disord 14, S194–S198.
Winn P, Wilson DIG & Redgrave P (2009). Subcortical connections of the basal ganglia. In Handbook of Basal Ganglia Structure and Function: A Decade of Progress, pp. 397–408, eds. Steiner H & Tseng K-Y. Academic Press (Elsevier), San Diego.