
Physiology News Magazine

Sleep disturbance alters autonomic balance to the heart
Sleep disturbances such as sleep apnoea can cause a chronic imbalance in the autonomic nervous system, and if left untreated, can lead to cardiovascular diseases.
Features
Sleep disturbance alters autonomic balance to the heart
Sleep disturbances such as sleep apnoea can cause a chronic imbalance in the autonomic nervous system, and if left untreated, can lead to cardiovascular diseases.
Features
Jhansi Dyavanapalli, Heather Jameson, Olga Dergacheva, Mona Alhusayyen & David Mendelowitz
Department of Pharmacology & Physiology, George Washington University, USA
Vivek Jain
Department of Medicine, George Washington University, USA
https://doi.org/10.36866/pn.97.28
Sleep disturbances, including obstructive sleep apnoea (OSA), are very common causes of cardiovascular diseases. For example, OSA, prevalent in ~24% of adult males and ~9% of adult females (between 30 and 60 years of age) (Young et al. 1993), is characterized by chronic repetitive interruptions of breathing during sleep. These episodic periods with suspensions of breathing (apnoea) cause cycles of combined hypoxia
(low oxygen) and increases in CO2 levels (hypercapnia) that often brings the person to a lighter state of sleep or brief wakefulness to stimulate and restore normal breathing. Left untreated, OSA may generate, and/or maintain, many cardiovascular diseases, including hypertension, arrhythmias and sudden cardiac death.
One of the key consequences of OSA is a chronic imbalance in the autonomic nervous system, which serves to maintain homeostatic cardiovascular function as well as to protect against challenges and perturbations to the cardiovascular system. Both the sympathetic and parasympathetic divisions of the autonomic nervous system regulate neural control of the heart, with parasympathetic activity dominating this balance. A normal resting heart rate of ~60–80 beats/minute is maintained by the tonic parasympathetic activity to the heart, without which heart rate would become elevated (~100 bpm) with a higher risk of arrhythmias. While previous work by others has focused on how OSA elicits sympathetic overactivity, the goal of the current study was to study and identify the mechanisms responsible for diminished cardioprotective parasympathetic control of heart rate in OSA. To accomplish these goals we utilised an animal model of chronic intermittent hypoxia–hypercapnia (CIHH) that mimics OSA in humans, and examined the critical synaptic pathways to neurons that generate the parasympathetic activity to the heart.
Parasympathetic cardiac vagal neurons (CVNs) of the brainstem, located in nucleus ambiguus and dorsal motor nucleus of the vagus, generate the parasympathetic activity to the heart. The vagal descending projections from these neurons synapse upon postganglionic intracardiac ganglia neurons located in close proximity to the heart (Mendelowitz, 1999). CVNs are intrinsically silent and hence their activity is dictated by the excitatory (glutamatergic) and inhibitory (GABA and glycinergic) synaptic activity arising from other regions of the brain (Mendelowitz, 1996) (Fig. 1).
A functional deficit in the brainstem circuitry responsible for parasympathetic activity to the heart (Gu et al. 2007), especially activation of CVNs (rather than anatomical or functional changes in peripheral innervation of the heart (Lin et al. 2007; Yan et al. 2008)) is responsible for impaired parasympathetic control of the heart with chronic intermittent hypoxia (CIH).
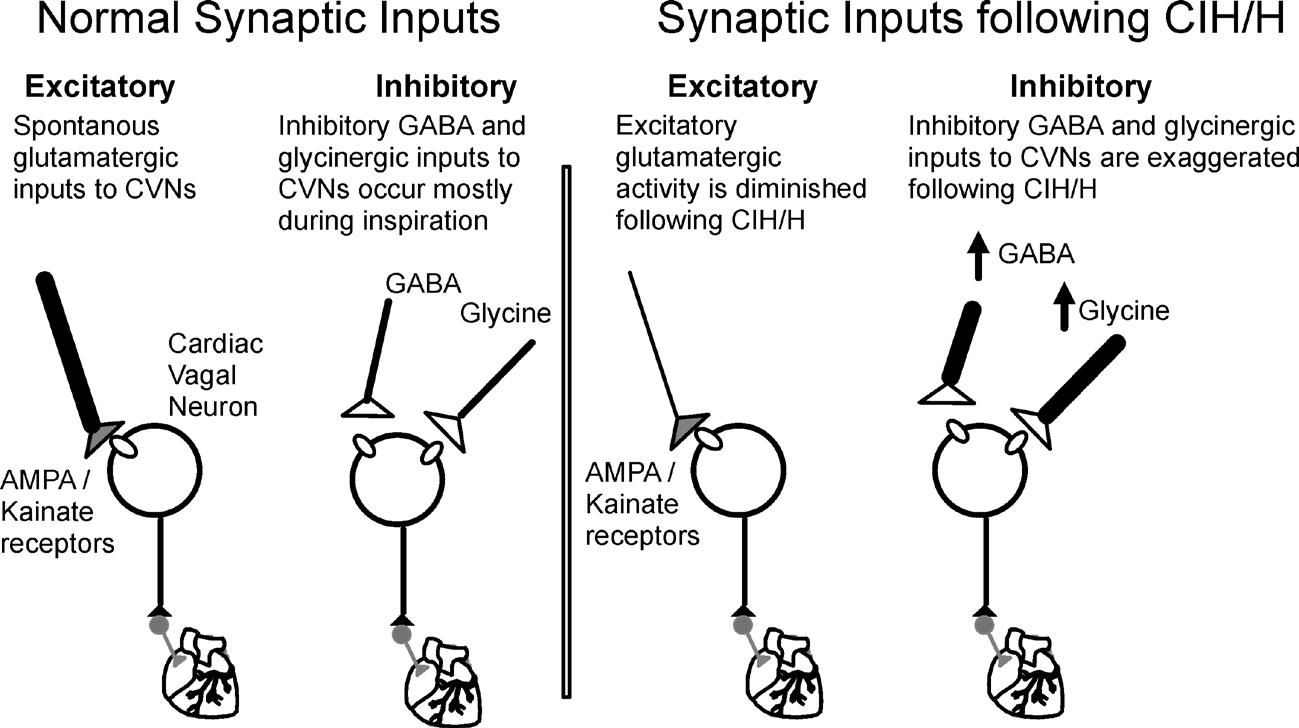
Supporting the hypothesis that CIH impairs CVN function in the brainstem, the heart rate response evoked upon microinjection of glutamate (Yan et al. 2008), into the nucleus ambiguus, where CVNs are located, is diminished by CIH. However, beyond alterations in glutamate receptor density little was known about how CIH impairs CVN function and what, if any, targets can be identified to restore cardioprotective parasympathetic activity to the heart.
The changes responsible for diminished vagal control of heart rate were identified by studying the changes in blood pressure, heart rate and neurotransmission to CVNs evoked by acute hypoxia–hypercapnia and CIHH. The methods have been described in detail in (Dyavanapalli et al. 2014). Briefly, in vivo telemetry recordings of blood pressure and heart rate were obtained in adult rats prior to and during 4 weeks of CIHH exposure. CIHH exposures were performed by placing the rats in commercial atmosphere controlled chambers, exposed to repetitive cycles of 3 minutes of mild hypoxia–hypercapnia (6% O2 + 5% CO2 + 89% N2) followed by 3 minutes of normoxia (21% O2 + 79% N2), repeated for 10 times/h, 8 h/day, for 4 weeks during the light phase. Fluorescently labelled CVNs were visualised and identified in an in vitro brainstem slice preparation obtained from adult rats exposed to either air or CIHH for 4 weeks. Neurotransmission to CVNs, including postsynaptic inhibitory and excitatory synaptic events, were recorded using whole cell voltage clamp techniques.
Our results have shown that chronic intermittent hypoxia–hypercapnia exposure for 4 weeks increases blood pressure to hypertensive levels and blunts cardiovascular reflexes in response to both acute hypoxia–hypercapnia and CIHH. This likely occurs by the observed changes in the neurotransmission to cardiac vagal neurons that normally maintain a low resting heart rate. Specifically, cardiac vagal neurons received an increased frequency of inhibitory (both GABA & glycinergic) and depressed incidence of excitatory (glutamatergic) neurotransmission with CIHH. These changes would act in concert to inhibit the activity of CVNs and diminish cardioprotective parasympathetic activity to the heart. These alterations of cardiorespiratory network function within the brainstem would contribute to the increased heart rate, blood pressure and risk of adverse cardiovascular events that occur in patients with OSA.
In addition to identifying the adverse alterations responsible for reduced parasympathetic activity to the heart following CIHH, the results from this study would predict that patients who have OSA and take sleep promoting medicines that typically act by enhancing inhibitory GABAergic neurotransmission within the CNS might be at heightened risk for a more significant reduction of critical parasympathetic neuronal activity to the heart. This study also provides a foundation for the development of potential therapeutic interventions to restore cardioprotective parasympathetic activity to the heart in patients with OSA.
References
Dyavanapalli J, Jameson H, Dergacheva O, Jain V, Alhusayyen M & Mendelowitz D (2014). J Physiol 592, 2799–2811.
Gu H, Lin M, Liu J, Gozal D, Scrogin KE, Wurster R, Chapleau MW, Ma X & Cheng ZJ (2007). Am J Physiol Heart Circ Physiol 293, H2809–2818.
Lin M, Liu R, Gozal D, Wead WB, Chapleau MW, Wurster R & Cheng ZJ (2007). Am J Physiol Heart Circ Physiol 293, H997–1006.
Mendelowitz D (1996). Am J Physiol 271, H2609–2614.
Mendelowitz D (1999). News Physiol Sci 14, 155–161.
Yan B, Soukhova-O’Hare GK, Li L, Lin Y, Gozal D, Wead WB, Wurster RD & Cheng ZJ (2008). Neuroscience 153, 709–720.
Young T, Palta M, Dempsey J, Skatrud J, Weber S & Badr S (1993). N Engl J Med 328, 1230-1235.