
Physiology News Magazine

Substrates and triggers for the initiation of arrhythmias
The mechanisms by which the spread of electrical waves through the heart often becomes disturbed in patients with electrical and/or structural heart diseases, leading to cardiac rhythm disturbances, remain incompletely understood. We exposed a novel mechanism for arrhythmia initiation, in which areas of abrupt cardiac tissue expansion and alterations in the inward sodium current responsible for cardiac excitation set the stage for spontaneous premature local re-excitation and arrhythmogenesis.
Features
Substrates and triggers for the initiation of arrhythmias
The mechanisms by which the spread of electrical waves through the heart often becomes disturbed in patients with electrical and/or structural heart diseases, leading to cardiac rhythm disturbances, remain incompletely understood. We exposed a novel mechanism for arrhythmia initiation, in which areas of abrupt cardiac tissue expansion and alterations in the inward sodium current responsible for cardiac excitation set the stage for spontaneous premature local re-excitation and arrhythmogenesis.
Features
David S. Auerbach and José Jalife
Center for Arrhythmia Research, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, USA
https://doi.org/10.36866/pn.85.15


Cardiac fibrillation
Cardiac electrical and structural heterogeneities set the stage for the initiation of complex cardiac arrhythmias, including atrial fibrillation (AF), which is the major cause of embolic stroke, and ventricular tachycardia/fibrillation (VT/VF), which is the largest immediate cause of sudden cardiac death (SCD). However, little is known about how alterations in the morphological structure and the electrical organization of the myocardium act in concert to result in the initiation of either AF or VT/VF. Causal abnormalities may include: altered expression of proteins that form and/or modulate membrane ion channel function; remodelling of intercellular electrical and mechanical junctions; and pathological changes in the extracellular matrix with the development of fibrosis.
Patients with acquired heart diseases such as ischaemic heart disease and heart failure are the most susceptible to arrhythmias due to impairment of the normal spread of electrical waves. In these patients, altered ion channel function and structural remodelling leading to fibrosis result in substantial electrical and structural heterogeneity. However, it is unknown which combination of factors contribute to the sudden appearance of AF or VT/VF. Patients who suffer from inherited cardiac diseases such as channelopathies and hypertrophic cardiomyopathy also are highly susceptible to arrhythmias and have an increased risk of embolic stroke and SCD. A case in point is long QT syndrome (LQTS), an inheritable channelopathy with an electrocardiographic phenotype of QT-interval prolongation (corrected QT-interval, QTc > 440 ms). These patients present with syncope, arrhythmogenesis (particularly torsades de pointes) and SCD, with the greatest prevalence during rest. Acquired LQTS can be manifested pharmacologically, or by electrolyte disturbances (hypokalaemia and hypomagnesaemia), structural heart disease or bradycardia. Many such patients are free of arrhythmias until one day the necessary conditions arise that lead to the onset of either AF or VT/VF. A great deal of research has been invested in understanding the conditions that initiate and maintain such arrhythmias, in an effort to develop novel strategies to prevent them.
Our recent study published in The Journal of Physiology (Auerbach et al. 2011) focused upon elucidating the substrate(s) and trigger(s) for the initiation of lethal arrhythmias. Specifically, in the presence of heterogeneous cardiac tissue architecture, we investigated the implications that alterations in the balance between depolarizing and repolarizing membrane ionic currents have upon the initiation of arrhythmias.
Previous work on arrhythmogenesis focused on anatomical or functional re-entry. However, little attention has been given to the possible role of the phenomenon of ‘reflection’ as a mechanism for the initiation and maintenance of arrhythmias. Reflection occurs when an electrical impulse that propagates along a narrow pathway returns spontaneously and prematurely along the same pathway, leading to extra beats and arrhythmia initiation. Reflection may occur if the pathway contains an area of impaired conduction (Antzelevitch et al. 1980) or a gradient in ion channel expression (Maoz et al. 2009). In our study, we demonstrated that reflection may depend on a structural heterogeneity consisting of a region of tissue expansion. A thin region of viable tissue that connected two wide regions of tissue promoted a transient local imbalance between inward and outward currents, prolongation of the action potential (AP) plateau, triggered activity (early after-depolarization, EAD), premature excitations and arrhythmogenesis. Most importantly, the possibility of reflection was significantly enhanced when structural defects combined with increased late or persistent sodium current (INa), such as seen in inherited and acquired cardiac electrical diseases.
Heterogeneous cardiac tissue architecture
The heart is a very heterogeneous structure. Its muscle has varying wall thickness, anisotropic fibre orientation, microvasculature and trabeculation (Fig. 1A), all of which impact the dynamics of impulse propagation. These heterogeneities exist in structurally normal cardiac tissue, including the sinoatrial node, the atrioventricular node and the Purkinje–ventricular muscle junction (Fig. 1B). Also, heterogeneities are further exacerbated by pathological conditions, such as accessory pathways (e.g. Wolf–Parkinson–White Syndrome), ischaemia, infarction and fibrosis (e.g. arrhythmogenic right ventricular cardiomyopathy/dysplasia). As shown in Fig. 1C, narrow pathways of myocytes, surrounded by fibrotic or necrotic tissue, or inflammatory cells, often branch and merge, leading to tortuous, fragmented and unstable electrical impulse propagation.

Our recent study provides a novel mechanism for the initiation of arrhythmias, whereby abrupt geometrical expansions provide a substrate for re-excitation and reflection (Fig. 1D–F).
Balance between depolarizing and repolarizing currents
The functional expression and biophysical properties of ion channels give the AP its characteristic shape. The INa and L-type calcium current (ICaL) are the two major depolarizing forces, while several potassium channel currents (Ito, IKr, IKs, IK1) serve to repolarize the cell. The balance between depolarizing and repolarizing currents determines the level of excitability, AP morphology, and the dynamics of normal and re-entrant impulse propagation. When there is a reduction or altered kinetics in the depolarizing INa, there is a concomitant increase in the susceptibility for conduction slowing, block and arrhythmogenesis, particularly at regions of geometrical expansion.
Therefore, strategies to increase INa density might serve as a potential anti-arrhythmic strategy. Recently Lau et al. (2009) used adenoviral transfer to express the skeletal muscle isoform of the sodium channel in the epicardial infarct border zone of the ventricle. In this zone there was an increase in the AP upstroke velocity, preserved fast conduction and a reduction in the incidence of inducible sustained VT/VF.
However, it is important to note that, when the depolarizing currents flowing during the AP plateau outcompete the repolarizing currents, for example, in LQTS type 3, a number of inherited mutations in the alpha subunit of the sodium channel result in the inability of INa to completely shut off during the AP. This results in the persistence of an inward current that contributes to prolongation of the AP plateau, with an increased susceptibility for triggered activity (EADs), premature excitation and arrhythmogenesis. Due to the great contributions of molecular biology, we have gained insights into the pathological remodelling in acquired cardiac disease (e.g. heart failure), as well as identified multiple inherited ion channel mutations. Patients with acquired and inherited ion channel diseases are vulnerable to SCD. For instance, genetic screening has implicated channelopathies as a cause of sudden infant death syndrome and sudden unexplained death in epilepsy.
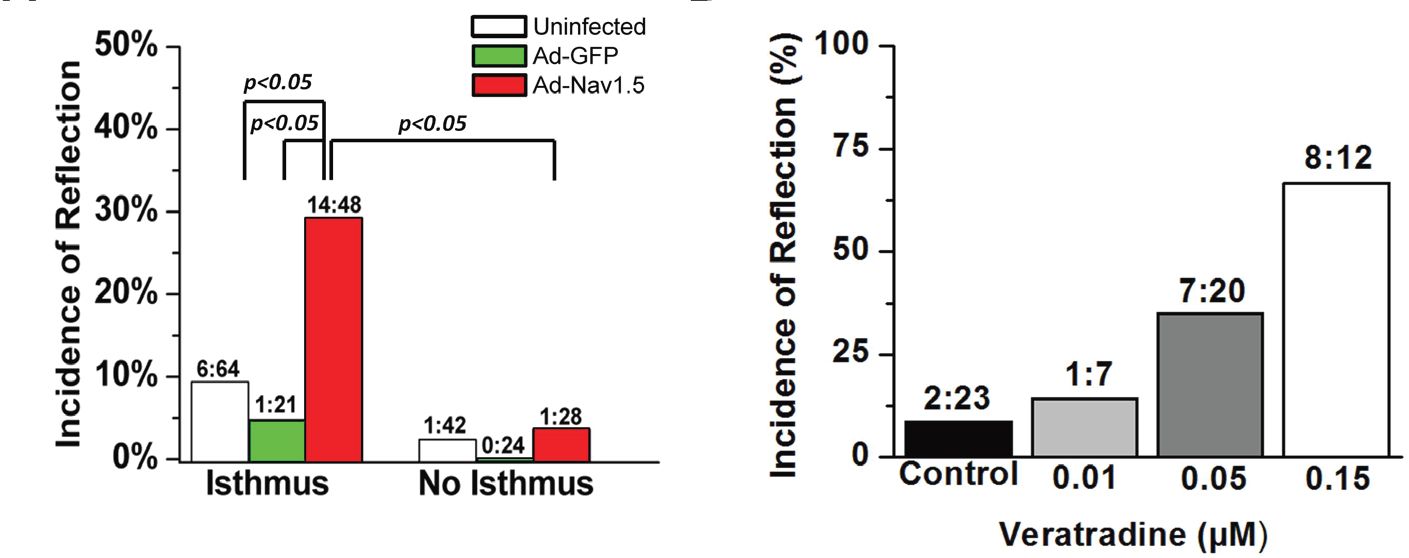
Advancing the understanding of arrhythmogenesis
In our study, adenovirally- and pharmacologically-increased persistent INa led to prolongation of the AP duration, EADs and reflection (Fig. 2), thus serving to model LQTS type 3.
A substrate (structural hetero geneity) and a trigger (increased persistent INa) combined to promote life-threatening arrhythmia initiation. As illustrated in Fig. 3, the abrupt geometrical expansion provided ideal conditions for retrograde electrotonic flow of depolarizing current into a region of high input resistance (i.e. thin strand of tissue) at a time of high membrane resistance (i.e. AP plateau).
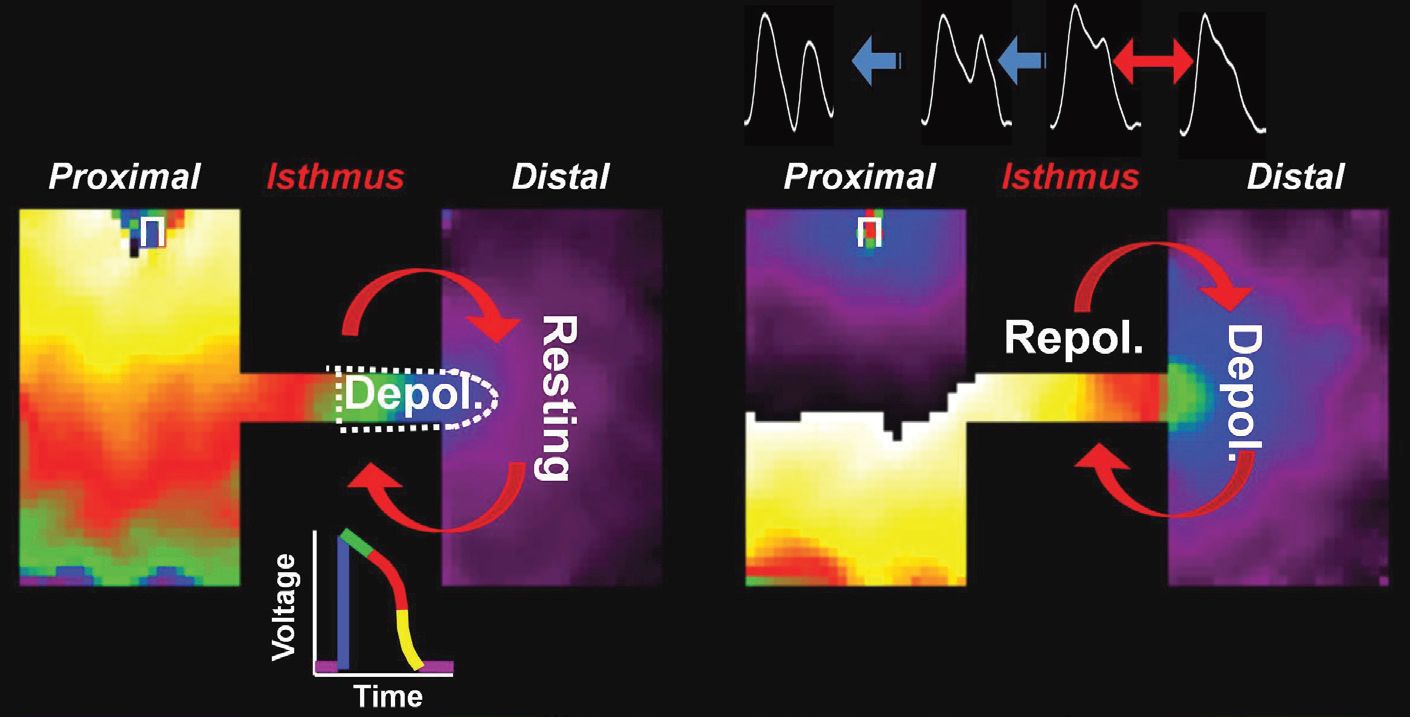
Our study provides a potential explanation for why patients with LQTS may be free of arrhythmias for many years until external factors (e.g. changes in the autonomic input to the heart, electrolyte imbalance, increased fibrosis, etc.) force the relation between substrate and trigger to reach a critical level at which time reflection, EADs and premature excitation occur. In patients with LQTS, coronary artery disease is an independent and significant risk factor increasing the incidence of LQTS-related cardiac events (Sze et al. 2008).
In conclusion, our model provides a new framework to examine the role that cardiac structure and alterations in ion channel expression and function have upon arrhythmogenesis. Results from our study offer insights into which patients may be at the greatest risk of arrhythmias initiated by reflection, and may subsequently provide clues towards targeted therapies to prevent the onset of VF.
References
Antzelevitch C, Jalife J & Moe GK (1980). Characteristics of reflection as a mechanism of reentrant arrhythmias and its relationship to parasystole. Circulation 61, 182–191.
Auerbach DS, Grzeda KR, Furspan PB, Sato PY, Mironov S & Jalife J (2011). Structural heterogeneity promotes triggered activity, reflection and arrhythmogenesis in cardiomyocyte monolayers. J Physiol 589, 2363–2381.http://jp.physoc.org/content/589/9/2363.full
Lau DH, Clausen C, Sosunov EA, Shlapakova IN, Anyukhovsky EP, Danilo P Jr et al. (2009). Epicardial border zone overexpression
of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation 119, 19–27.
Maoz A, Krogh-Madsen T & Christini DJ (2009). Instability in action potential morphology underlies phase 2 reentry: a mathematical modeling study. Heart Rhythm 6, 813–822.
Sze E, Moss AJ, Goldenberg I, McNitt S, Jons C, Zareba W, Qi M & Robinson JL (2008). Long QT syndrome in patients over 40 years of age: increased risk for LQTS-related cardiac events in patients with coronary disease. Ann Noninvasive Electrocardiol 13, 327–331.