
Physiology News Magazine

The diversity of sex development
What do conditions affecting sex development teach us about sexual diversity?
Features
The diversity of sex development
What do conditions affecting sex development teach us about sexual diversity?
Features
https://doi.org/10.36866/pn.123.16
Dr Angela Lucas-Herald, University of Glasgow, UK
Professor Syed Faisal Ahmed, University of Glasgow, UK
Approximately 1 in 50 newborns will be born with a structural or functional change in their body, often referred to as a congenital anomaly. Congenital anomalies, also known as birth defects, may actually be even more common than this, with some not presenting until a later stage in life. These conditions can have a variable effect on the newborn’s health. In Europe, in approximately 1 in 1,000 cases, the infant may not survive the first year of life (Boyle et al., 2018). However, in many cases, the congenital anomaly may not have a major direct impact on the child’s health but could simply be a signal for a wider group of conditions that may have long-term effects. Lastly, there may also be other relatively minor isolated congenital anomalies that do not have any impact on the child’s health. A clear knowledge of the cause of the congenital anomaly as well as information on condition-specific long-term outcome allows a carer to reach a plan that is personalised for the needs of a specific infant.
Atypical genitalia are a relatively common form of congenital anomaly and may affect approximately 21 in 10,000 cases worldwide (Yu et al., 2019). In around three-quarters of these cases, the affected child is a boy (Ahmed et al., 1999, Ahmed et al., 2004). Although this presentation is rarely linked to life-threatening conditions in the newborn, parents and healthcare staff find the birth of the infant with atypical genitalia challenging especially when the genitalia are so atypical that the sex cannot be assigned immediately. Although these situations are, strictly speaking, not medical emergencies, the premium that society places on issues related to sex development and sex assignment, means that an immediate response is required even if it is aimed at reassurance and explanation by an expert.
Recent data from Scotland show that delayed sex assignment because of a concern related to external genitalia occurs in less than 1 in 10,000 infants (Rodie et al., 2019) and with this level of rarity, it is no wonder that the public as well as healthcare staff find these situations so challenging. In some societies, delayed sex assignment at an older age in childhood or the option to not assign sex are accepted alternatives but it remains unclear as to whether these alternatives lead to lower levels of physical or psychosocial morbidity.
Normal and atypical sex development
Over the last few decades, a greater understanding of the developmental biology of the sex organs has allowed a deeper insight into the conditions that may affect sex development. In mammals (as shown in Fig. 1) sex development occurs in two distinct and sequential stages. The first stage relates to the development of the gonads and the second stage relates to the development of the genitalia, both internal and external. Gonadal development is determined by the complement of sex chromosomes and leads to the specific development of either a male or female gonad from a single undifferentiated bipotential gonad.

The presence of a Y chromosome drives the bipotential gonad toward testis-specific differentiation, whereas its absence results in development of an ovary (Lucas-Herald and Bashamboo, 2014). Increasingly it is clear that there are other factors that can act as anti-testis factors and can push the pathway in the developing testis towards an ovary. There are also pro-ovarian factors, deficiency of which can reduce the potential for ovarian development. A reverse in gonadal development to what might be expected is rarely so complete that a male-specific gonad is replaced by a perfectly functioning female-specific gonad or vice versa. In most cases, gonadal development is disrupted to such an extent that the gonad degenerates at some stage in life, its ability to produce sex hormones or eggs is limited or it may pose a tumorigenic risk (Andrade et al., 2019). Very rarely, in possibly less than 1 in 100,000 births, an infant may possess normally functioning testis and ovarian tissue within a single gonad (Caputo et al., 2019).
The production of androgens such as testosterone and dihydrotestosterone and other hormones such as anti-Müllerian hormone (AMH) by the testis results in the development of the male-typical internal and external genitalia (prostate, vas deferens, penis and scrotum) with a reciprocal regression of the Müllerian ducts, which are the precursors of the female-typical internal genital structures (Fallopian tubes, uterus and upper vagina) (Rey and Grinspon, 2011). However, a deficiency in the production or action of androgens in an XY infant can lead to a reduction in the androgenisation of the male genitalia whilst an excess in an XX infant can lead to androgenisation of the female genitalia. Similarly, a deficiency in the production or action of AMH in an XY infant can lead to the presence of a uterus in a boy with almost male-typical external genitalia (Picard et al., 2017).
In boys, atypical genitalia commonly present as undescended testes, hypospadias, small phallus or a combination of these. A quarter of children with these conditions have an additional congenital anomaly in another organ system and around a third may have an endocrine or genetic condition that affects their gonadal function (Cox et al., 2014). Furthermore, many of these boys may also have adverse long-term health outcomes and the likelihood of these may depend on the underlying endocrine or genetic diagnosis. So, whilst in the past, it was often thought that reconstruction of the genitalia would lead to a resolution of the condition, it is becoming increasingly clear that in some cases, these conditions may have a greater significance, which will only become clearer with detailed longitudinal follow
up of physical and mental health outcomes.
Genetics versus hormones
Differences between boys and girls, men and women are not just manifested by differences in physical characteristics but also in the sexually dimorphic nature of several behavioural traits as well as predisposition to illnesses. It is possible that some of these are conditioned by sociocultural factors. However, male humans are more likely to develop bacterial infections and women are more likely to develop autoimmunity. Furthermore, many forms of neurological and neurocognitive conditions including Alzheimer’s disease, Parkinson’s disease, autism, addiction, depression, anxiety disorders and schizophrenia show a clear sex difference and there is good evidence to support that both genetics and the gonadal hormonal milieu may both have independent effects in modulating this sexual specificity (Dunn et al., 2011).
The idea that there is more to sex differences than hormones was first described in the studies performed on the zebra finch almost 30 years ago when hormonal manipulation in the male or female bird did not alter the genetic male’s ability to sing his distinct courtship song (Arnold, 1997). It is also possible that some genes that play a critical role in gonadal development and, thus, sex hormone synthesis play a direct role through hormone-independent pathways. For instance, SRY, the gene on the Y chromosome that directs the bipotential mammalian gonad to develop as testes is also expressed in the human foetal adrenal, brain, heart and pancreas as well as the heart, kidney and liver in the adult.
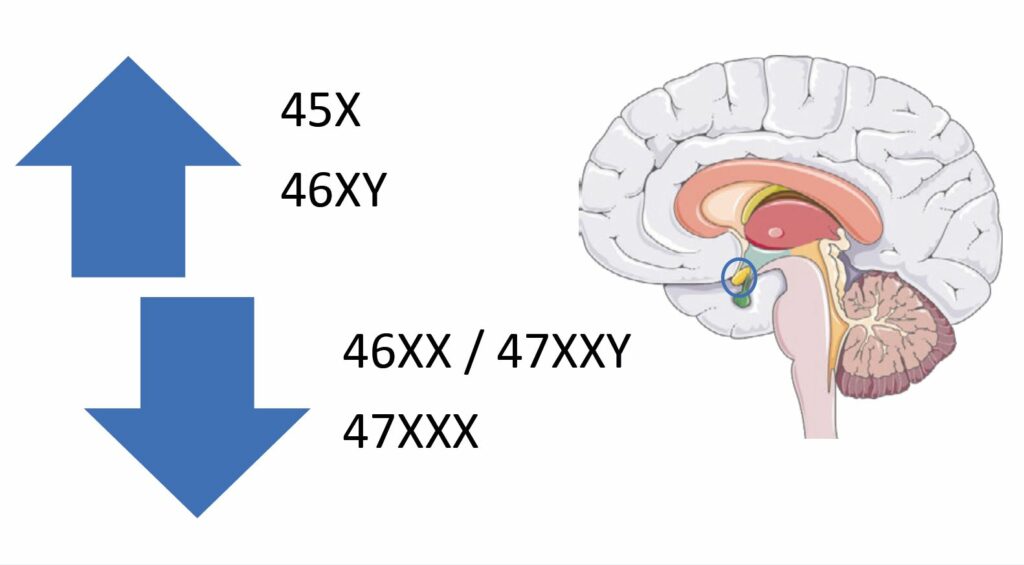
Lastly, there is also good evidence that there are some sex chromosome-linked genes that may also contribute to the sexual dimorphism. For instance, on the X chromosome, there are genes, such as monoamine oxidase A & B, that are reported to play a dosage-sensitive role in modulating the serotonergic pathway and the development of the amygdala, a part of the brain’s limbic system critical for the expression of conditioned fear (Reardon, 2016). In humans, the size of the amygdala is reported to be inversely related to the number of X chromosomes (Fig. 2), such that women with Turner syndrome who have a 45X karyotype have a larger amygdala than 46XX women, and men with Klinefelter syndrome, who have a 47XXY karyotype, have an amygdala that is smaller than 46XY men and similar to 46XX women. In accordance with this, women who are 47XXX have a smaller amygdala than 46XX women (Reardon et al., 2016). However, the size of the amygdala and its activity may also be modulated by sex hormones as evident from functional imaging studies across the menstrual cycle as well as in pathological conditions associated with excess androgen exposure.
Disorder of sex development
The phrase disorder of sex development (DSD) was coined in 2005 to move away from terms such as intersex, which may have different meanings to different people, or pseudohermaphroditism (which is a nightmare to type or pronounce!). Some feel that the term “disorder” in DSD is too pathologising and reinforces the need for correction and would prefer the word “difference”. One could relate this paradigm to statural height, which is normally distributed. However, the further away somebody is from the mean, their height becomes abnormal and when they are a few standard deviations away from the mean, then their abnormal height may also be associated with a functional disability. In addition, in some people abnormal height may, itself, not be disabling but may act as a sign of other pathology. Congenital heart anomalies are usually described as congenital heart disease (CHD); one would think that the word “disease” may have an even stronger connotation of morbidity than a disorder, but it is well accepted in that field that some heart conditions that are included within the category of CHD do not need any intervention. Thus, the stance to take could be that atypical genitalia confirm that sex development has progressed in a different way to usual and that there is a need for further investigation to exclude an underlying disorder that may or may not require therapy.
Conditions that are associated with a DSD also cast a spotlight on society’s views on the anatomical features that a body should possess for being male or female. XY boys who have AMH deficiency (persistent Müllerian duct syndrome) will have a uterus (Picard et al., 2017). And then there are girls and women with a condition called androgen insensitivity syndrome who have an XY karyotype and testes but no uterus. There are also conditions, such as 5 alpha- reductase deficiency, where the young XY child has female typical external genitalia until the age of puberty after which the external genitalia become more male typical.
Lastly, DSDs also teach us important lessons on differences between the development of gender identity and sex development. Gender identity is the individual’s perception of one’s characteristics and how they may socially conform to the norms, behaviours and roles that may be typically associated with being a boy, girl, man or a woman. Clearly, a newborn child, irrespective of whether they have a DSD or not can be assigned a sex at birth but there is an assumption that the child’s gender development will be in accordance with the assigned sex. Whilst there is a possibility that the child may identify with another gender at a later age, studies suggest that the likelihood of this is very low. Recent studies suggest that, overall, dissatisfaction with one’s own gender in people with a past history of DSD is similar to the background population (Kreukels et al., 2018). However, there are some specific conditions where gender dysphoria may be more common, and usually in these conditions, prenatal exposure to androgens may be a contributory factor (Hines et al., 2015).
Conclusion
So, whilst the books tell us that biological development into male and female individuals usually follows typical pathways, there are several examples from nature that illustrate the point that, in reality, there is a lot more to sexual diversity.
References
Ahmed SF et al. (1999). Assessment of the gonadotrophin–gonadal axis in androgen insensitivity syndrome. Archives of Disease in Childhood 80, 324- 329. http://doi.org/10.1136/adc.80.4.324
Ahmed SF et al. (2004). Prevalence of hypospadias and other genital anomalies among singleton births, 1988–1997, in Scotland. Archives of Disease in Childhood – Fetal and Neonatal Edition 89, F149-F151. http://doi.org/10.1136/adc.2002.024034
Andrade JGR et al. (2019). Clinical findings and follow- up of 46,XY and 45,X/46,XY testicular dysgenesis. Sexual Development 13, 171-177. http://doi.org/10.1159/000504239
Arnold AP (1997). Sexual differentiation of the zebra finch song system: positive evidence, negative evidence, null hypotheses, and a paradigm shift. Journal of Neurobiology 33, 572-584. http://doi.org/10.1002/(SICI)1097-4695(19971105)33:5<572::AID-NEU6>3.0.CO;2-1
Boyle B et al. (2018). Estimating global burden of disease due to congenital anomaly: an analysis of European data. Archives of Disease in Childhood – Fetal and Neonatal Edition 103, F22. http://doi.org/10.1136/archdischild-2016-311845
Caputo M et al. (2019). Ovotesticular disorder of sex development: A rare case of lateral subtype 45X/46XY karyotype diagnosed in adulthood. Urology 129, 68- 70. http://doi.org/10.1016/j.urology.2019.04.008
Cox K et al. (2014). Novel associations in disorders of sex development: findings from the I-DSD Registry. The Journal of Clinical Endocrinology and Metabolism 99, E348-55. http://doi.org/10.1210/jc.2013-2918
Dunn GA et al. (2011). Sex-specificity in transgenerational epigenetic programming. Hormones and Behavior 59, 290-295. http://doi.org/10.1016/j.yhbeh.2010.05.004
Hines M et al. (2015). Early androgen exposure and human gender development. Biology of Sex Differences 6, 3. http://doi.org/10.1186/s13293-015-0022-1
Kreukels BP et al. (2018). Gender dysphoria and gender change in disorders of sex development/intersex conditions: results from the dsd-LIFE study. The Journal of Sexual Medicine 15, 777-785. http://doi.org/10.1016/j.jsxm.2018.02.021
Lucas-Herald, AK and Bashamboo A (2014). Gonadal development. Understanding Differences and Disorders of Sex Development (DSD). Karger Publishers, Basel, Switzerland.
Picard JY et al. (2017). The persistent Müllerian Duct Syndrome: An update based upon a personal experience of 157 cases. Sexual Development 11, 109-125. http://doi.org/10.1159/000475516
Reardon PK et al. (2016). An allometric analysis of sex and sex chromosome dosage effects on subcortical anatomy in humans. Journal of Neuroscience 36, 2438-2448. http://doi.org/10.1523/jneurosci.3195-15.2016
Rey RA and Grinspon RP (2011). Normal male sexual differentiation and aetiology of disorders of sex development. Best Practice & Research Clinical Endocrinology & Metabolism 25, 221-238. http://doi.org/10.1016/j.beem.2010.08.013
Rodie M et al. (2019). Nationwide study of the prevalence and initial management of atypical genitalia & delayed sex assignment in the newborn. 58th Annual ESPE, 2019. European Society for Paediatric Endocrinology.
Yu X et al. (2019). Hypospadias prevalence and trends in international birth defect surveillance systems, 1980–2010. European Urology 76, 482-490. http://doi.org/10.1016/j.eururo.2019.06.027