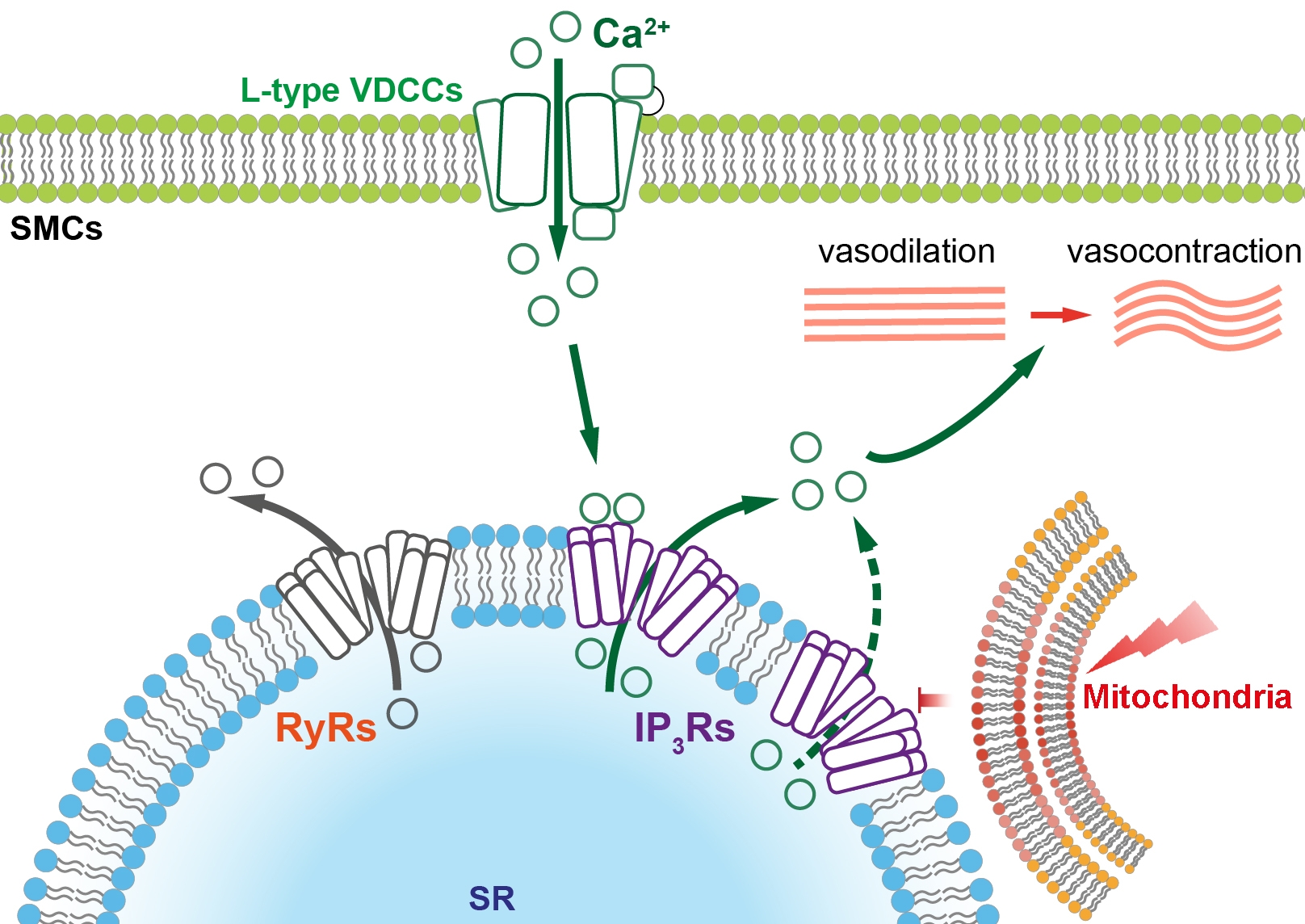
The contractility of vascular smooth muscle cells (VSMCs) in resistance arteries is the major contributor of vascular tone and blood pressure. In VSMCs, increase of intracellular Ca2+ level directly generates the force to contract arteries. A major source of intracellular Ca2+ is entry through voltage dependent Ca2+ channels (VDCCs) on plasma membrane. While mitochondria are now recognised as key regulators of intracellular Ca2+ homeostasis by modulating internal Ca2+ store in several cell types including smooth muscle cells and endothelial cells, their role in modulating Ca2+ signalling generated by VDCC in intact VSMCs is still under studied. Given the potential interaction between mitochondria and VDCCs in VSMCs, we hypothesize that mitochondria can directly regulate Ca2+ release from sarcoplasmic reticulum in VSMCs, hence regulate Ca2+ signalling and vascular tone generated mediated by VDCCs.
The interplay between mitochondria and VDCCs was investigated by imaging and analysing intracellular Ca2+ signals in smooth muscle cells in intact arteries from rat mesentery (n = 5). Summarized data were analyzed and presented as mean ± SD of n biological replicates. When the data extracted from same preparations under different treatment, data were analyzed by using paired t-test.
Depolarization of the plasma membrane potential, by high potassium (30 mM) physiological saline solution, triggered Ca2+ entry through VDCCs and a sustained increase in intracellular Ca2+ on which repetitive Ca2+ oscillations occurred. All Ca2+ signals were abolished by removal of external Ca2+ and by dihydropyridine inhibitors of VDCCs. Significantly, the repetitive Ca2+ oscillations, but not the sustained Ca2+ signals, were blocked by the IP3 receptor inhibitor 2-APB and SERCA inhibitor cyclopiazonic acid. Neither the repetitive Ca2+ oscillations or the sustained Ca2+ signals were altered by the ryanodine receptor inhibitors ryanodine and dantrolene (paired t-test). These results suggest that Ca2+ entry via VDCC triggers Ca2+-induced Ca2+ release via IP3 receptors in intact mesenteric arteries.
Depolarization of the mitochondrial membrane potential (ψm, indicated by a membrane sensitive dye, TMRE), by the uncoupler CCCP (1 µM), but not ATP deprivation with the ATP synthesis blocker oligomycin (1 µM), inhibited VDCCs evoked IP3 mediated Ca2+ oscillations but not the sustained Ca2+ signals. Furthermore, in intact arteries, depolarization of ψm directly suppressed inositol triphosphate receptors (IP3Rs) mediated Ca2+ release from the internal Ca2+ store evoked by photolysis of caged IP3. These results suggest that mitochondria regulate Ca2+ release via IP3Rs triggered by voltage dependent Ca2+ entry. In return, Ca2+ entry via VDCCs did not alter ψm. In addition, depolarization of ψm supressed sustained vasocontraction that was mediated by voltage dependent Ca2+ entry.
Together, these results suggest that mitochondria regulate Ca2+-induced Ca2+ release at IP3 receptors triggered by Ca2+ entry via VDCCs but do not directly regulate VDCCs activity.