
Physiology News Magazine
Our hyper-polarizing story: understanding ATP-mediated vasodilatation in humans
Negative results, cynicism, the piecing together of puzzles and integrating physiology with experimental approaches. It’s all part of the inspiring and meandering journey on the path to understand ATP-mediated vasodilatation.
Features
Our hyper-polarizing story: understanding ATP-mediated vasodilatation in humans
Negative results, cynicism, the piecing together of puzzles and integrating physiology with experimental approaches. It’s all part of the inspiring and meandering journey on the path to understand ATP-mediated vasodilatation.
Features
Anne Crecelius
University of Dayton, USA
Frank Dinenno
Colorado State University, USA
https://doi.org/10.36866/pn.94.38
While it is satisfying to be ‘right’ as scientists, we know sometimes the most important progress occurs when data are surprising or contrary to our original hypothesis. The pursuit of explanations for unexpected findings often leads to the best advancements. The story of how our laboratory came to investigate the underlying vasodilator pathways of adenosine triphosphate (ATP) (Crecelius et al. 2012) is an example of such a path to our current understanding.
Regulation of vascular tone and the endothelium
Since the early 20th century, physiologists have been interested in skeletal muscle blood flow regulation, particularly during occasions of stress to the body, such as muscle contraction (exercise) or changes in gravitational force (standing). Eventually it was realized the endothelium, a monolayer of epithelial cells separating the lumen of the vessel from the smooth muscle cells, was an important mediator of vascular tone and capable of releasing factors that elicited vasodilatation. Robert F. Furchgott first posed the idea of an ‘endothelium-derived relaxing factor’ (EDRF) (Furchgott & Zawadzki, 1980), later identified by Furchgott and Louis J. Ignarro as the gaseous nitric oxide (NO).
In 1998, a Nobel Prize was awarded for the discoveries concerning NO as a cardiovascular signalling molecule. We and others were aware of the potential relationship between NO and cardiovascular health, particularly as it related to ageing. Initially, we focused on the impact of age and regular aerobic exercise training on large artery structure and function, conduit and resistance vessel endothelial function, and peripheral blood flow under resting conditions. Many of these studies did not address NO specifically; however, bioavailability of this molecule, either attenuated with age or improved with exercise, was a proposed mechanism to a number of our observations.
Eventually, we used intra-arterial pharmacology to address mechanisms of local regulation of vascular tone and performed a variety of studies, many to determine the role of NO by utilizing exogenous L-arginine analogues (the precursor for endothelial nitric oxide synthase (NOS)-derived NO) to inhibit NO synthesis. We also possessed the ability to directly stimulate and inhibit the action of the sympathetic nervous system with specific receptor agonist and antagonists, fuelling our rapid growth of understanding how the nervous system influences skeletal muscle blood flow.
As the new millennium was ushered in, the phenomenon of ‘functional sympatholysis’ (see box), first proposed by Remensmyder in the early 1960s (Remensnyder et al. 1962), was observable in both human and animal studies in a variety of preparations. Regarding underlying mechanisms of this response, Thomas and colleagues demonstrated that functional sympatholysis in the contracting rat hindlimb was impaired in the presence of NOS inhibition (Thomas & Victor, 1998). However, when we attempted to translate these findings of a significant role for NO in functional sympatholysis to our human studies, we were unable to do so. We took a variety of approaches, but continually failed to observe a significant ability for NO to blunt sympathetically mediated vasoconstriction when given directly as sodium nitroprusside, as well as observed intact functional sympatholysis in the presence of NOS inhibition (Dinenno & Joyner, 2003). Additionally, NO did not appear to be obligatory for vasodilatation during muscle contractions and combined inhibition of NO and other endothelial-derived vasodilators such as prostaglandins (PGs) resulted in a modest decline (~25%) in exercise hyperaemia when inhibition occurred during muscle contractions (Schrage et al. 2004). Taken together, while evidence was strong for the vasoprotective properties of NO in humans, data from our laboratory and others indicated that vascular regulation during muscle contractions appeared to be somewhat independent of this endothelial-derived vasorelaxant.
Interestingly, the lack of an obligatory role for NO in both exercise hyperaemia and functional sympatholysis in humans was somewhat perplexing given our findings demonstrating impaired vasodilatation and functional sympatholysis in healthy, sedentary older individuals during exercise, a group well-characterized by decreased NO bioavailability (Kirby et al. 2010, 2011). Had we observed that NO was obligatory in these cases, a logical theory of impaired blood flow control during exercise in older adults would have existed. This was not the case, and it would take us several more years of both basic science and clinical studies before we formed an overall hypothesis for what molecule other than NO might explain these collective findings.
Extracellular ATP: unique vascular capabilities
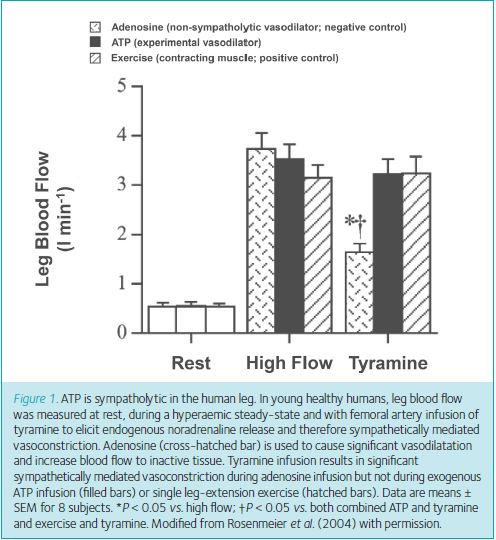
Early studies appreciated the diverse function of purines in cardiovascular regulation, but it was not until the late 1990s that investigators began to develop a hypothesis regarding the role of erythrocyte-released extracellular ATP as a mediator of the matching of muscle perfusion to oxygen demand (Ellsworth et al. 2009). Building upon this idea, in 2004 Rosenmeier and colleagues demonstrated that circulating ATP was capable of overriding sympathetic vasoconstriction in the leg evoked by the infusion of tyramine, a drug that elicits endogenous noradrenaline (NA) release (Fig. 1) (Rosenmeier et al. 2004). The finding of an exogenous substance capable of being ‘sympatholytic’ was rather surprising as over the years we had tested many substances in this regard, none of which mimicked what occurred in contracting skeletal muscle.
In truth, we were quite sceptical of these intriguing findings, and thus we designed a study in the forearm to confirm ATP was capable of overriding sympathetically mediated vasoconstriction and specifically address whether this occurred post-junctionally, as is the case during muscle contractions. The synaptic level of modulation was unable to be determined from the findings of Rosenmeier and colleagues given their experimental approach of tyramine-induced vasoconstriction. To our surprise, we built upon the results from the Scandinavian group and showed ATP is clearly capable of post-junctional modulation of sympathetically mediated vasoconstriction and importantly, that this occurs in a graded manner such that a low dose of exogenous ATP is not sympatholytic, whereas higher doses are (Kirby et al. 2008). The graded nature of the sympatholytic ability of ATP parallels the graded effect of exercise intensity on functional sympatholysis. We were then convinced that in addition to being a potent vasodilator, ATP possesses unique signalling properties that merit additional study.
At this time our interest in ageing began to merge with our developing knowledge of vasoactive ATP. Animal-based experiments indicated that ATP-mediated vasodilatation was dependent on an intact endothelium and this led us to pursue whether ATP could be used as a test of endothelial function in older adults, similar to how the muscarinic agonist acetylcholine was used by our group and others. However, contrary to our hypothesis, ATP-mediated vasodilatation was not impaired in older individuals, despite these subjects demonstrating ‘endothelial dysfunction’ as characterized by impaired dilator responses to acetylcholine (Kirby et al. 2010). This ‘incorrect’ hypothesis would be critical in the development of our future research that tried to explain this unanticipated finding. Firstly, what endothelial-derived vasodilators was ATP stimulating? Secondly, whatever this mechanism might be, could it lend important insight into the unexplained aspects of vascular regulation during exercise?
Several lines of evidence suggested that what was termed ‘endothelial dysfunction’ at that point could be more specifically termed ‘decreased NO bioavailability’. Given ATP seemed to be dissociated from NO bioavailability based on our dilatory study in older adults (Kirby et al. 2010) and the studies on sympatholysis in younger adults (Kirby et al. 2008), we pursued what vasodilator pathways were stimulated by exogenous ATP. Initially, we investigated the role of the endothelial-derived vasodilators NO and PGs, as the existing literature was equivocal in this regard. No matter the manner in which we tested the idea, at varied doses and timing of inhibition and multiple measurement techniques of muscle blood flow, we were unable to inhibit ATP-mediated vasodilatation more than ~20–30% (Crecelius et al. 2011). We were left to consider the non-NO, non-PG mechanisms of endothelium-dependent vasodilatation.
Endothelial-derived hyperpolarization: factoring it in
Upon closer examination of the literature from animal models and isolated vessel preparations, we began to appreciate that non-NO, non-PG mechanisms of vasodilatation were likely to be critical for vascular control at rest and in response to stimulation. However, similar to the original unknown nature of NO and generalized name of EDRF, these pathways were not well described and generally termed endothelial-derived hyperpolarizing factors (EDHFs); a response dependent on the endothelium that caused a characteristic hyperpolarization (decrease in membrane potential) of endothelial and vascular smooth muscle cells leading to vasodilatation (Feletou & Vanhoutte, 2007). One hypothesis proposed potassium (K+) could be an EDHF given its potential release from endothelial cells and ability to stimulate the Na+/K+-ATPase pump and inwardly rectifying potassium channels (KIR) on vascular smooth muscles causing membrane potential hyperpolarization and vasorelaxation (Edwards et al. 1998). An important aspect of the proposed mechanism for K+ was its efflux through calcium-activated K+ channels (KCa). Taken together with data that demonstrated inhibition of small- and intermediate-conductance KCa channels essentially abolished vasodilatation to luminal ATP application in the rat mesentery (Edwards et al. 1998), we had even more motivation to try and address hyperpolarizing mechanisms of vasodilatation in our human model as it might lend insight to our previous findings regarding ATP.
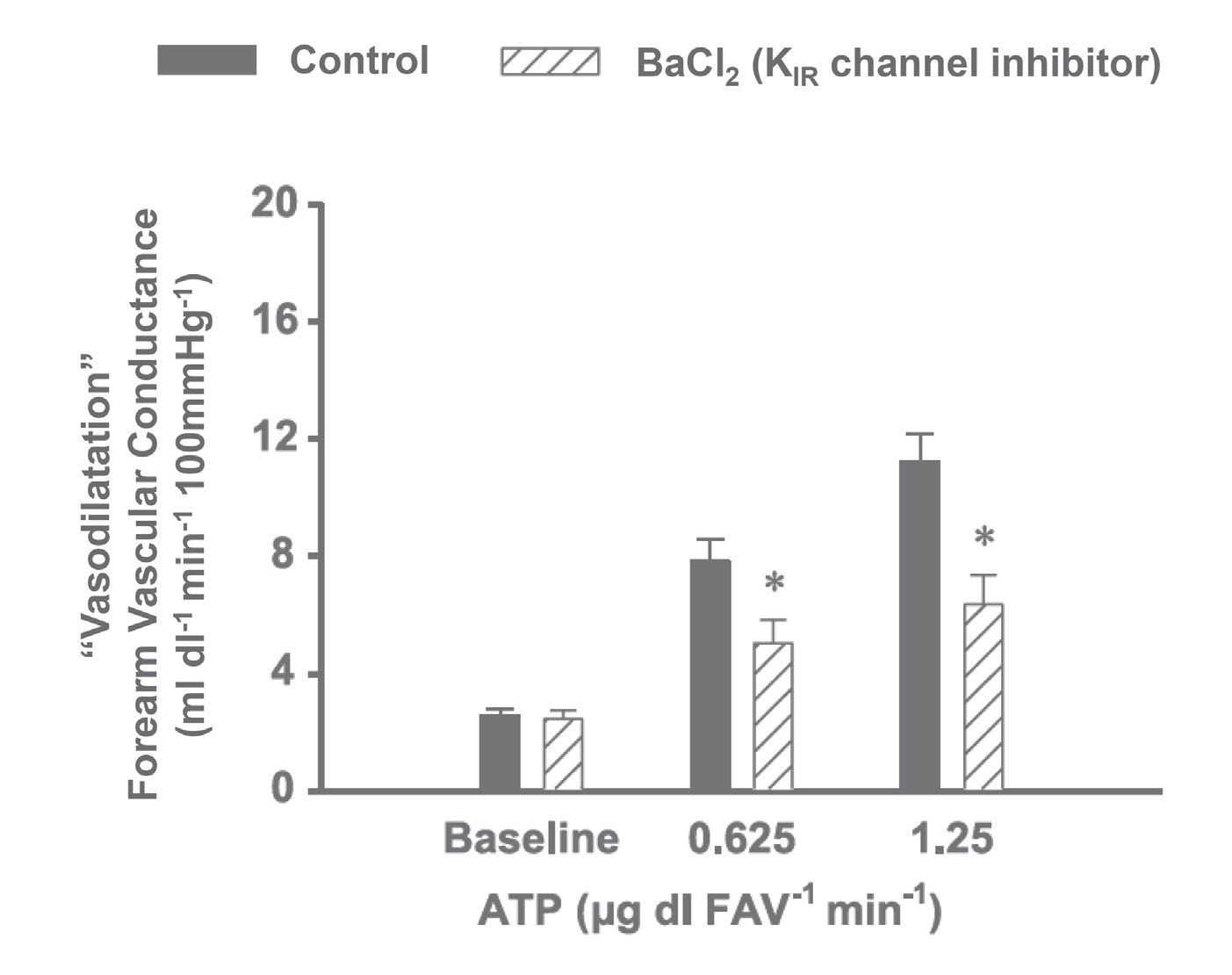
To date, investigators interested in inhibiting vascular hyperpolarization in humans were somewhat limited in the pharmacology that could be safely administered to do so. The toxins and venoms often used in isolated vessel preparations to inhibit small- and intermediated-conductance KCa channels posed safety risks. Enzymatic inhibition of specific proposed stimuli of hyperpolarization (e.g. cytochrome p450, H2O2, etc.) left readers questioning efficacy since directly challenging these inhibitors was difficult. We were excited to find that barium chloride (BaCl2) and ouabain could be given intra-arterially in low doses to inhibit KIR channels and Na+/K+-ATPase, respectively (Dawes et al. 2002). Importantly, low doses of KCl could be given intra-brachially to cause hyperpolarization and directly challenge inhibitor efficacy (Fig. 2). We saw an exciting opportunity to try and translate the isolated vessel experiments that supported a role for hyperpolarization in ATP-mediated vasodilatation (Winter & Dora, 2007) by inhibiting downstream mechanisms of VSMC hyperpolarization (Edwards et al. 1998).

The ‘wow’ moment
It was necessary to first establish our ability to sufficiently inhibit a hyperpolarizing stimulus (exogenous KCl) with the combination of BaCl2 and ouabain, as had previously been described (Dawes et al. 2002). With these control experiments completed (in fairly profound fashion as we essentially eliminated any K+-mediated vasodilatation), we began our first experiments to test the impact of inhibition of KIR channels and Na+/K+-ATPase on ATP-mediated vasodilatation (Crecelius et al. 2012). These first studies were some of the more exciting in the laboratory to date. Given our data are collected in real-time, we were able to immediately estimate the changes in muscle blood flow following inhibition. From the first subject forward, it was clear these inhibitors were more ‘powerful’ than those we had used before and a large portion of ATP-mediated vasodilatation was explained by these pathways. After a number of experimental approaches, we determined the primary mechanism of ATP-mediated vasodilatation is vascular hyperpolarization via activation of KIR channels (Fig. 3) (Crecelius et al. 2012).
Putting it all together
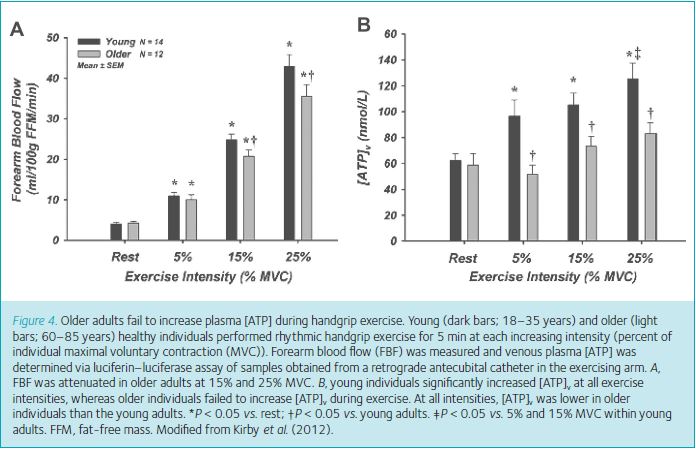
As we were working to understand the basic signalling mechanisms of ATP, we were concurrently studying the potential role for ATP in the control of vascular tone during exercise in young and older adults, given the finding in young subjects that in sufficient concentration, it was sympatholytic (Rosenmeier et al. 2004; Kirby et al. 2008). Interestingly, although not completely surprising given our emerging overall working hypothesis, we demonstrated ATP was capable of blunting direct sympathetically mediated vasoconstriction in older individuals, despite these individuals having impaired exercise-induced functional sympatholysis (Kirby et al. 2011). Had we dissociated ATP and sympatholysis with these findings? Or, was it possible ATP retained the same vascular signalling capabilities in older adults, but this population failed to increase ATP during exercise to the same concentration as young individuals? We had begun to make plasma measures of ATP in the laboratory and pursued this hypothesis to tie our multiple studies together. This time, our ageing hypothesis was ‘correct’ as we showed older individuals failed to increase plasma ATP during incremental handgrip exercise in contrast to the significant increase observed in younger individuals (Fig. 4) (Kirby et al. 2012). Thus, while we admittedly have not been able to directly inhibit the purinergic receptors to which ATP binds, as no pharmacology has been established that is capable of doing so, our findings of the mechanisms for ATP-mediated vasodilatation and our ageing data continue to support our overall hypothesis regarding a role for ATP in muscle blood flow regulation during exercise (Fig. 5).

Along the way
The ability to inhibit KIR channels and Na+/K+-ATPase has provided the laboratory with a powerful pharmacological tool that we continue to use to address a number of hypotheses. Armstrong and colleagues had demonstrated a role of K+-mediated vasodilatation in rapid vasodilatation following a single muscle contraction in the hamster cremaster (Armstrong et al. 2007) using BaCl2 and ouabain along with a pharmacological inhibitor of K+ efflux from skeletal muscle. Given our interest in rapid vasodilatation and the minimal number of mechanistic studies on the topic (Kirby et al. 2007), attempting to translate the findings of Armstrong and colleagues seemed prudent. In general, in line with the findings in the animal model, we demonstrated a significant role for K+ in mediating a portion of rapid vasodilatation in response to a single muscle contraction at a variety of intensities (Crecelius et al. 2013a).
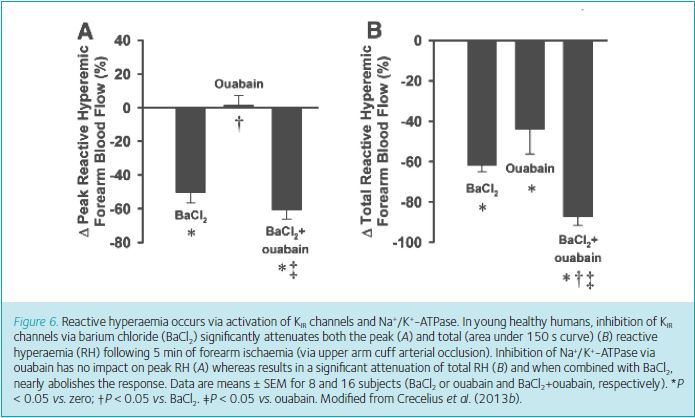
Of perhaps greater clinical relevance, we recently determined whether reactive hyperaemia (RH), the large increase in blood flow following release of temporary ischaemia of skeletal muscle, was mediated by KIR channels and Na+/K+-ATPase. Although RH is a useful test of microvascular function and has clinical value in assessing risk of cardiovascular health (Anderson et al. 2011), prior to our work the underlying signalling mechanisms were largely undetermined. Impressively, nearly all (~90%) of the total (area under the curve) RH can be explained by combined activation of KIR channels and Na+/K+-ATPase and ~50% of the peak response is due to KIR channel activation (Fig. 6) (Crecelius et al. 2013b). This magnitude of inhibition is by far the most profound that has been observed to date.
Moving forward
We continue to pursue our ideas regarding muscle blood flow regulation, specifically during exercise and the unique role ATP may have in modulating sympathetic vasoconstriction. It is often challenging to translate mechanistic findings from animal models and in vitro studies to our human model, given our concerns for human safety. Part of what we find inspiring from this story is the integration of physiology and experimental approaches that was required to reach the point we find ourselves at. We drew from findings in older individuals to guide our basic science studies as well as applying our findings from young healthy humans to older individuals. While we probably would not have pursued many of these ideas without the initial findings and discoveries related to NO, we have developed a cautious attitude towards ‘endothelial function’ as we have seen first-hand this monolayer is far more involved in vascular regulation than simply being a source of NO. Similarly, mechanistic studies in vitro and in animal models continue to stimulate ideas and provide rationale; yet, we observe important differences when translating some of these approaches to humans. Importantly, we acknowledge that scepticism, negative findings and unsupported hypotheses have provided us important directions we have excitedly followed.
References
Anderson TJ, Charbonneau F, Title LM, Buithieu J, Rose MS, Conradson H, Hildebrand K, Fung M, Verma S & Lonn EM (2011). Microvascular function predicts cardiovascular events in primary prevention: long-term results from the Firefighters and Their Endothelium (FATE) study. Circulation 123, 163–169.
Armstrong ML, Dua AK & Murrant CL (2007). Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581, 841–852.
Crecelius AR, Kirby BS, Luckasen GJ, Larson DG & Dinenno FA (2012). ATP-mediated vasodilatation occurs via activation of inwardly rectifying potassium channels in humans. J Physiol 590, 5349–5359.
Crecelius AR, Kirby BS, Luckasen GJ, Larson DG & Dinenno FA (2013a). Mechanisms of rapid vasodilation following a brief contraction in human skeletal muscle. Am J Physiol Heart Circ Physiol 305, H29–40.
Crecelius AR, Kirby BS, Richards JC, Garcia LJ, Voyles WF, Larson DG, Luckasen GJ & Dinenno FA (2011). Mechanisms of ATP-mediated vasodilation in humans: modest role for nitric oxide and vasodilating prostaglandins. Am J Physiol Heart Circ Physiol 301, H1302–H1310.
Crecelius AR, Richards JC, Luckasen GJ, Larson DG & Dinenno FA (2013b). Reactive hyperemia occurs via activation of inwardly rectifying potassium channels and Na+/K+-ATPase in humans. Circ Res 113, 1023–1032.
Dawes M, Sieniawska C, Delves T, Dwivedi R, Chowienczyk PJ & Ritter JM (2002). Barium reduces resting blood flow and inhibits potassium-induced vasodilation in the human forearm. Circulation 105, 1323–1328.
Dinenno FA & Joyner MJ (2003). Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: is nitric oxide obligatory? J Physiol 553, 281–292.
Dinenno FA, Masuki S & Joyner MJ (2005). Impaired modulation of sympathetic α-adrenergic vasoconstriction in contracting forearm muscle of ageing men. J Physiol 567, 311–321.
Edwards G, Dora KA, Gardener MJ, Garland CJ & Weston AH (1998). K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 396, 269–272.
Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH & Sprague RS (2009). Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology (Bethesda) 24, 107–116.
Feletou M & Vanhoutte PM (2007). Endothelium-dependent hyperpolarizations: past beliefs and present facts. Ann Med 39, 495–516.
Furchgott RF & Zawadzki JV (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376.
Jantzi MC, Brett SE, Jackson WF, Corteling R, Vigmond EJ & Welsh DG (2006). Inward rectifying potassium channels facilitate cell-to-cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol 291, H1319–1328.
Kirby BS, Carlson RE, Markwald RR, Voyles WF & Dinenno FA (2007). Mechanical influences on skeletal muscle vascular tone in humans: insight into contraction-induced rapid vasodilatation. J Physiol 583, 861–874.
Kirby BS, Crecelius AR, Voyles WF & Dinenno FA (2010). Vasodilatory responsiveness to adenosine triphosphate in ageing humans. J Physiol 588, 4017–4027.
Kirby BS, Crecelius AR, Voyles WF & Dinenno FA (2011). Modulation of postjunctional α-adrenergic vasoconstriction during exercise and exogenous ATP infusions in ageing humans. J Physiol 589, 2641–2653.
Kirby BS, Crecelius AR, Voyles WF & Dinenno FA (2012). Impaired skeletal muscle blood flow control with advancing age in humans: attenuated ATP release and local vasodilation during erythrocyte deoxygenation. Circ Res 111, 220–230.
Kirby BS, Voyles WF, Carlson RE & Dinenno FA (2008). Graded sympatholytic effect of exogenous ATP on postjunctional α-adrenergic vasoconstriction in the human forearm: implications for vascular control in contracting muscle. J Physiol 586, 4305–4316.
Remensnyder JP, Mitchell JH & Sarnoff SJ (1962). Functional sympatholysis during muscular activity. Observations on influence of carotid sinus on oxygen uptake. Circ Res 11, 370–380.
Rosenmeier JB, Hansen J & Gonzalez-Alonso J (2004). Circulating ATP-induced vasodilatation overrides sympathetic vasoconstrictor activity in human skeletal muscle. J Physiol 558, 351–365.
Schrage WG, Joyner MJ & Dinenno FA (2004). Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol 557, 599–611.
Thomas GD & Victor RG (1998). Nitric oxide mediates contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol 506, 817–826.
Winter P & Dora KA (2007). Spreading dilatation to luminal perfusion of ATP and UTP in rat isolated small mesenteric arteries. J Physiol 582, 335–347.