
Physiology News Magazine
Store-operated calcium entry in adult skeletal muscle fibres: the missing clue
Features
Store-operated calcium entry in adult skeletal muscle fibres: the missing clue
Features
Bruno Allard & Vincent Jacquemond
Physiologie Intégrative, Cellulaire et Moléculaire, Université Lyon 1, CNRS, Villeurbanne, France.
https://doi.org/10.36866/pn.66.26

Changes in cytoplasmic Ca2+ concentration are of ubiquitous importance to the function of living cells. A rise in cytoplasmic [Ca2+] can occur from either Ca2+ entry through the plasma membrane or Ca2+ release from an intracellular store, mainly the endoplasmic reticulum. One obvious difference between the two mechanisms is that the store has a finite content and will, to some extent, get depleted upon substantial activation of a Ca2+ release process. In the past 20 years considerable evidence has been accumulated supporting the existence of a specific plasma membrane Ca2+ entry pathway meant to refill emptied (or partially emptied) Ca2+ stores. Although there is sparse indication that, in certain cell systems, a direct pathway between the extracellular medium and the lumen of the store could do the job, the most widely accepted view is that the way is through the cytoplasm. Then, store depletion-induced Ca2+ entry generates a cytoplasmic [Ca2+] elevation, the role of which goes in fact well beyond replenishment of the store as it appears to be strongly relevant for various cell signalling processes. The study of ‘store operated calcium entry’ has become an extensive field of research, covered recently by a number of comprehensive reviews (see, for instance, Parekh & Putney, 2005).

The adult skeletal muscle fibre is a robust archetype of a cell relying on an intracellular Ca2+ store for its function: Ca2+ release from the sarcoplasmic reticulum (SR) is activated by membrane depolarization and generates the cytoplasmic [Ca2+] elevation that triggers contraction, whereas Ca2+ entry through the plasma membrane is not relevant to this process (see Melzer et al. 1995). Actually even the most well known Ca2+ entry pathway in skeletal muscle (through the voltage-dependent slow Ca2+ channels) has no clear established function. Still, and although the store-operated Ca2+ entry concept is mostly popular in non-excitable cells, the possibility was there too, that SR Ca2+ depletion would trigger a specific Ca2+ influx through the plasma membrane of skeletal muscle fibres. This was supported by a series of experimental data by Kurebayashi & Ogawa (2001), showing in particular that the rate of Mn2+ entry into muscle fibres (measured by a fluorescence quenching approach) was increased when the SR was severely depleted. This study was an encouraging piece of work, prompting further analysis of the physiological properties of storeoperated Ca2+ entry in muscle. The interest of this quest further gained in intensity when it was suggested that dysfunction of store-operated channels belonging to the TRPC (transient receptor potential canonical) family was responsible for an abnormal, potentially detrimental, influx of Ca2+ in dystrophin-deficient muscle (Vandebrouck et al. 2002). One could then put a name to an ion channel protein with an identified gating mechanism in relation to the dystrophic pathology. Interestingly, the unitary activity of the corresponding ion channel appeared well resolvable with the cell-attached patch-clamp technique and actually was very similar to a single channel activity previously shown to be particularly high in dystrophin-deficient cells by several groups (see Gillis, 1999). This is important because in many other instances, store-operated calcium entry was proven easier to reveal with fluorescent methods than with membrane current measurements. At that point we thought that a further indepth characterization of the store dependent Ca2+ influx in control and dystrophin-deficient muscle fibres could bring precious information into the field. More specifically, we were very interested in the possibility of directly measuring the store-dependent membrane current under acute conditions of SR Ca2+ depletion, in an intact muscle fibre under voltageclamp. This latter condition is of critical importance as membrane polarization is a non-avoidable parameter if one wishes to study the physiological properties of either transplasma membrane or trans-SR membrane Ca2+ fluxes in muscle cells, and thus a fortiori when studying a possible relationships between the two, as is the case for an SR-operated Ca2+ entry mechanism. Since voltage-clamp had so far not been used in previous studies of store-operated Ca2+ entry in muscle fibres, we thought this may be a key to a wide, unexplored aspect of the problem. We worked things out trying to gather the best possible sets of experimental conditions so that we would not miss the store-operated Ca2+ current. This included, on the same muscle fibre, a combination of either two or three techniques: whole-cell voltage-clamp (allowing control of SR Ca2+ release and measurement of the total membrane current), cell-attached patch-clamp (for tracking the abovementioned store-dependent-suspected unitary channel activity) and detection of the fluorescence of a calcium dye injected within the cytoplasm of the fibre. Well, despite our efforts, all this failed! We tested a vast array of experimental protocols and conditions aimed at depleting the SR such as repetitive large membrane depolarizations, use of SR Ca2+ uptake blocking agents or of SR Ca2+ releasing agents, but never detected any reproducible change in either unitary or whole-cell membrane current that would be consistent with a store-operated Ca2+ influx (Allard et al. 2006).
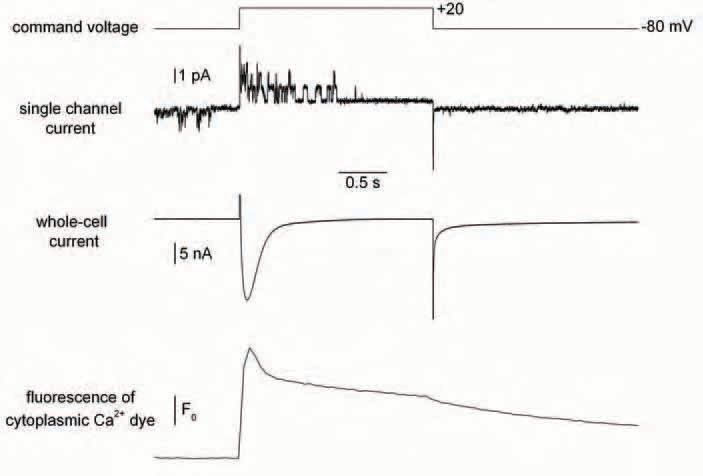
Now why is that? How could it be that the Ca2+ influx would not show up under these conditions, when SR Ca2+ depletion was assessed and membrane current measured at both the macroscopic and unitary levels? Why could not we detect any sign of an inward current? One could speculate that, for obscure reasons, SR contentdependent Ca2+ entry did not operate under this specific set of conditions. Conversely we tend to believe that assessing the existence of a storedepletion induced Ca2+ influx in a muscle fibre out of voltage control is quite hazardous. Another possibility would be to consider that the influx is too small to be detected as a current with electrical methods whereas it would be detectable with fluorescent quenching techniques. Still, then what would be the physiological relevance of this non-electrically detectable current when, for instance, we know that there is a clear well-detected Ca2+ entry through the voltage–dependent Ca2+ channels upon membrane depolarization, the role of which is unknown. This makes the physiological relevance of this putative store-operated Ca2+ entry very much questionable. There are several other detailed aspects of the problem as well that may be discussed but overall it seems that in the absence of new related experimental data the controversy will stand.
Acknowledgements
The preparation of this manuscript and the related work done in our laboratory were supported by grants from Université Lyon 1, Centre National de la Recherche Scientifique and Association Française contre les Myopathies.
References
Allard B, Couchoux H, Pouvreau S & Jacquemond V (2006). Sarcoplasmic reticulum Ca2+ release and depletion fail to affect sarcolemmal ion channel activity in mouse skeletal muscle. J Physiol 575, 69-81.
Gillis JM (1999). Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J Muscle Res Cell Motil 20, 605-625.
Kurebayashi N & Ogawa Y (2001). Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J Physiol 533, 185-199.
Melzer W, Herrmann-Frank A Lüttgau HC (1995). The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim Biophys Acta 1241, 59-116.
Parekh AB & Putney JW (2005). Store-operated calcium channels. Physiol Rev 85, 757-810.
Vandebrouck C, Martin D, Colson-Van Schoor M, Debaix H & Gailly P (2002). Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol 158, 1089-1096.