Introduction
Chronic kidney disease (CKD) drives adverse cardiac remodeling, characterized by interstitial fibrosis, diastolic dysfunction, and increased arrhythmic susceptibility, even in early disease stages. Early-stage CKD is associated with a six-fold elevation in sudden cardiac death risk, predominantly due to ventricular arrhythmia. A hallmark of CKD is persistent sympathetic nervous system (SNS) over activation, which perturbs cardiac electrophysiological and mechanical properties. However, the mechanistic link between renal dysfunction, SNS dysregulation, and ventricular arrhythmogenesis remains incompletely understood.
Aim
To investigate CKD-induced SNS dysregulation and its contribution to ventricular arrhythmia susceptibility in murine hearts.
Materials and Methods
Female C57BL/6J mice were fed either 0.15% adenine (n = 11) or control chow (n = 13) for 7 weeks to induce CKD. Renal dysfunction was quantified using serum BUN, creatinine, and normalised kidney weight. Cardiac electrophysiology was assessed via in vivo ECG and ex vivo optical mapping, with ventricular arrhythmia susceptibility evaluated using programmed stimulation. SNS activity was determined by heart rate variability (HRV) and circulating noradrenaline levels (ELISA). Cardiac structure and blood pressure were assessed by echocardiography and haemodynamic measurements. Results are presented as control versus adenine.
Results
Adenine-fed mice demonstrated increased serum BUN (8.5±0.9mmol/L vs 23.4±6.0mmol/L, p<0.0001), elevated creatinine (14.6±3.7umol/L vs 39.4±23.9umol/L, p<0.0001), and reduced kidney weight (251.8±26.90mg/g vs 188.6±33.9mg/g, p<0.0001), confirming renal impairment.
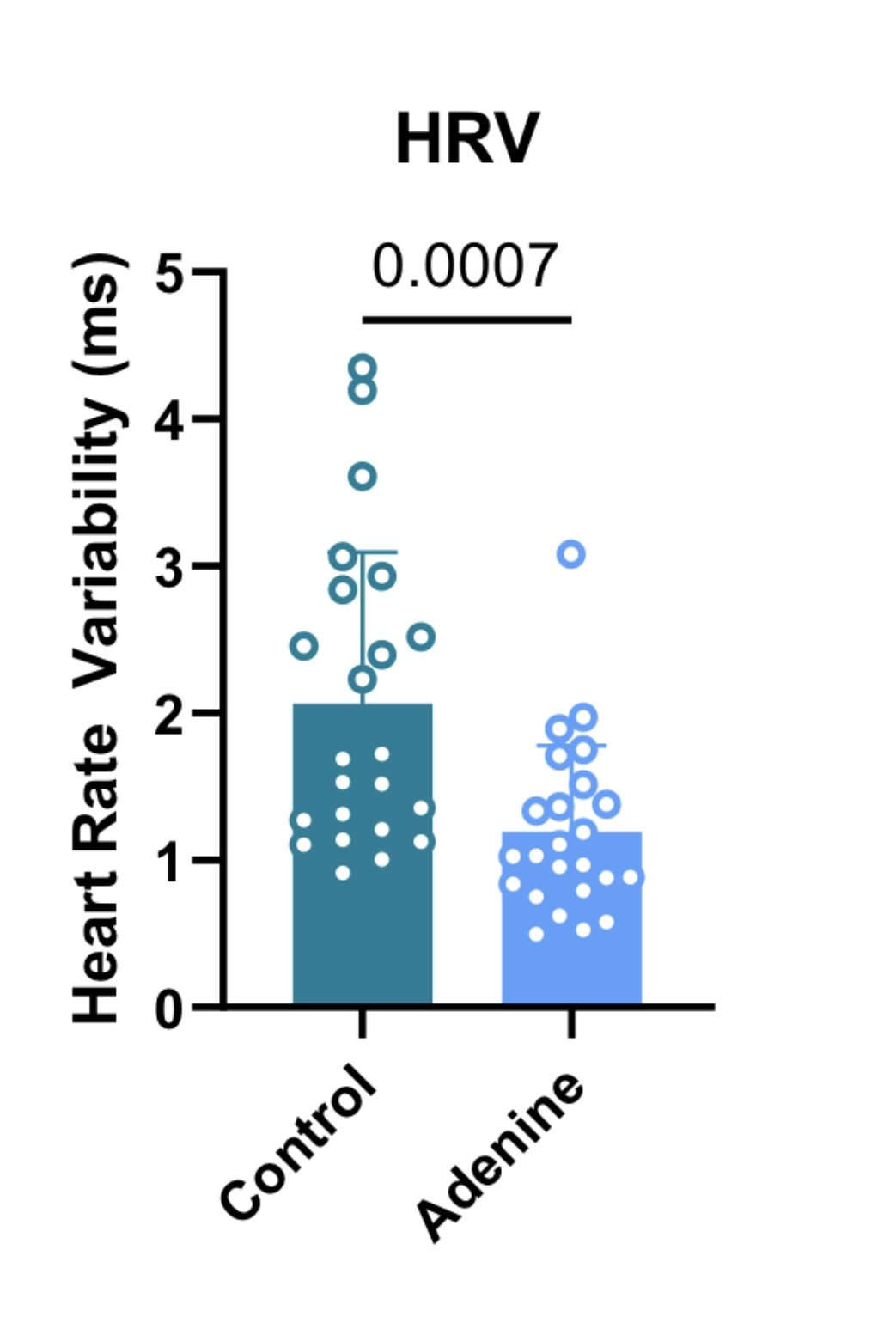
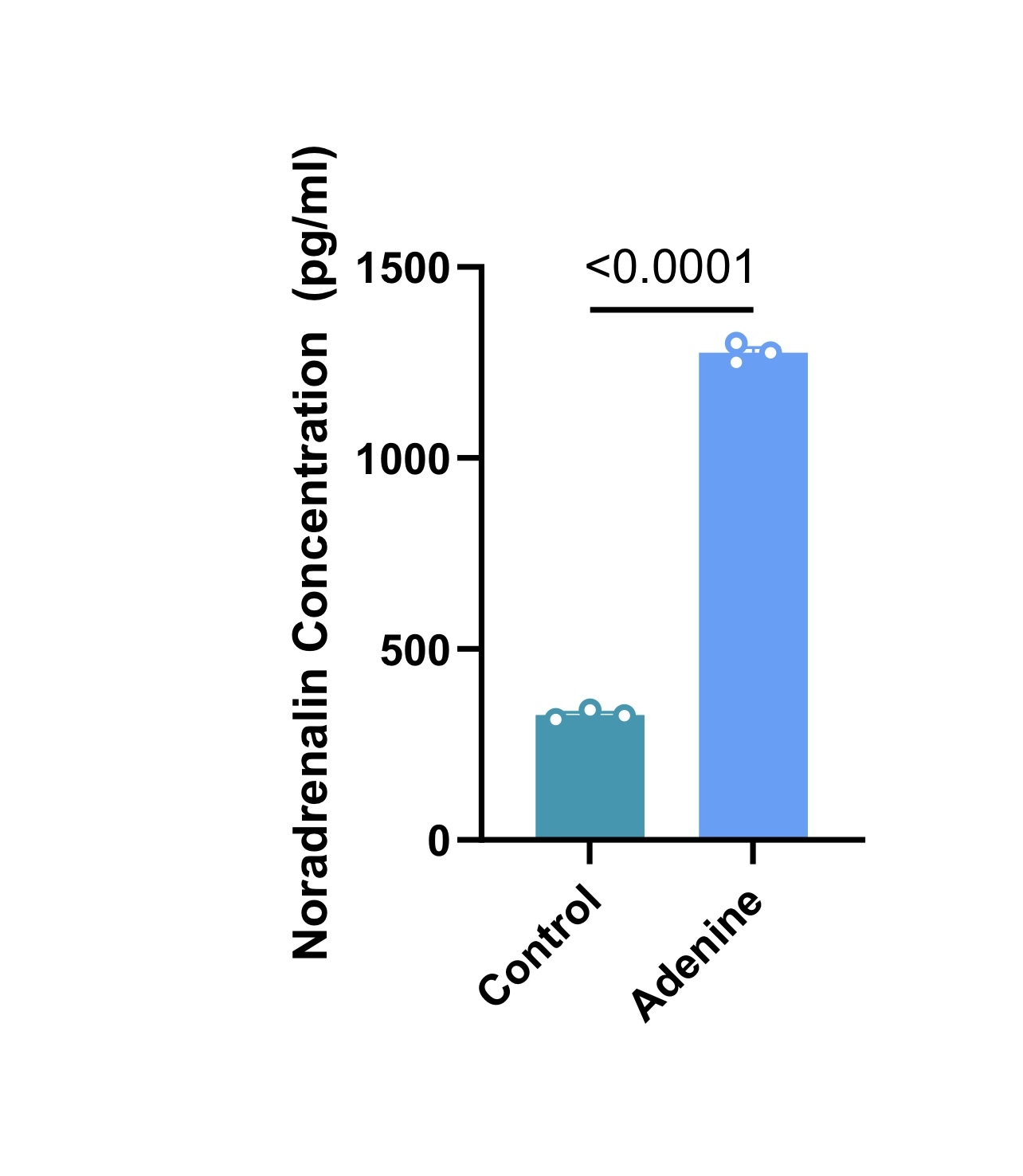
Adenine diet reduced HRV (figure 1) (2.1±1.0ms vs 1.2±0.6ms, p = 0.0007) and markedly elevated circulating noradrenaline (figure 2) (326.7±12.6pg/ml vs 1275±25pg/ml, p < 0.0001), despite reduced heart rate (736.6±22.56bpm vs 703.4±33.0bpm, p=0.0002), suggesting impaired chronotropic responsiveness and autonomic dysfunction.
Electrophysiological alterations included P-duration shortening (12.3±0.7 vs 11.2±0.9ms, p=0.0007) and PR-interval prolongation (33.0±2.0ms vs 35.3±2.5ms, p=0.0014). Ex vivo optical mapping showed action potential duration prolongation. For example, at 120ms pacing cycle length, there was a significant action potential duration prolongation at 30% (14.4±1.6 vs 18.7±3.9, p=0.002), 50% (27.6±4.9 vs 35.6±8.2, p=0.006) and 80% repolarisation (49.6±6.7 vs 58.4±12.4, p=0.04).
Furthermore, adenine mice displayed amplitude alternans, indicating beat-to-beat electrical instability, across pacing cycle lengths (p <0.0001). 58% of adenine hearts displayed pacing-induced arrythmias, primarily ventricular tachycardia (75%) and ventricular fibrillation (25%), compared to 0% of control. These changes occurred without overt structural remodelling or hypertension.
Conclusion
Adenine-induced CKD in mice leads to significant SNS dysregulation, alongside bradycardia, PR prolongation, prolonged action potential duration, electrical instability, and increased ventricular arrhythmias. The coexistence of sympathetic activation with bradycardia suggests impaired autonomic control and neuro-cardiac remodelling contributing to arrhythmogenesis.